
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Well-defined polyacrylamides with AIE properties via rapid Cu-mediated living radical polymerization in aqueous solution: thermoresponsive nanoparticles for bioimaging†
Congkai
Ma
a,
Ting
Han
b,
Niu
Niu
b,
Lucas
Al-Shok
a,
Spyridon
Efstathiou
 a,
Daniel
Lester
c,
Steven
Huband
d and
David
Haddleton
*a
a,
Daniel
Lester
c,
Steven
Huband
d and
David
Haddleton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk
bCenter for AIE Research, College of Materials Science and Engineering, Shenzhen University, Shenzhen, 518060, China
cPolymer Characterisation Research Technology Platform, University of Warwick, Coventry, CV4 7AL, UK
dDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
First published on 2nd December 2021
Abstract
There is a requirement for the development of methods for the preparation of well-controlled polymers with aggregation-induced emission (AIE) properties. This requirement directed this current work towards a robust synthetic route, which would be applicable for preparation in water and the presence of many types of functional groups. Herein, aqueous Cu-mediated living radical polymerization (LRP) has been optimized to provide facile and rapid access to a diverse range of water-soluble AIE polymers at sub-ambient temperatures. Homo-, block and statistical copolymerization all proceeded to a near full monomer conversion (≥99%) within 1 or 2 h and exhibited narrow dispersity, even when DP was targeted up to 1000. This excellent control associated with this polymerisation technique and the high-end group fidelity achieved were further demonstrated by linear first order kinetics and successful in situ block copolymerization, respectively. Fine-tuning the monomer sequence and composition of poly(N-isopropylacrylamide) (PNIPAM) copolymers allows for different lower critical solution temperature (LCST) and fluorescent thermoresponsive nanoparticles, which spontaneously self-assembled to varying sizes in water as determined by a combination of techniques (DLS, SAXS and TEM). Additionally, the fluorescence intensity was demonstrated to depend on the polymer concentration, architecture of the side chains and temperature. Particularly, PNIPAM-containing polymers were resistant to reduction in thermo-induced emission. The good biocompatibility, photostability and high specificity make them promising candidates as lysosome-specific probes for application in bioimaging.
Introduction
Until the concept of aggregation-induced emission (AIE) was coined by Tang and coworkers in 2001,1 molecular aggregation had been regarded to be detrimental to luminescence due to the aggregation-caused quenching (ACQ) phenomenon. Intriguingly, the AIE effect has revolutionarily demonstrated that aggregation can allow for boosted light emission with an appropriate chemical moiety, contributing potential application in optoelectronics,2–4 environmental detection and monitoring,5–7 as well as biomedical areas8–12 over the last decade. Notably, the expansion of AIE research into polymer science enables the fabrication and synthesis of different functional luminescent materials, which can benefit from the excellent mechanical properties of polymers and ready regulation of their composition, structure, and morphology.13 To date, the majority of AIE-active polymers have been prepared by click polymerization,14–17 free radical polymerization,18 ring-opening polymerization,19 and polycoupling20 mediated by one-, two- or multi-component polymerization. This has furnished many complex polymeric materials with high atom economy and diverse structures.21Nevertheless, there is still a requirement to develop facile, robust and rapid synthetic strategies to enable the efficient construction of AIE polymers with monomer sequence control and targeted molecular weights as well as well-defined architectures under mild conditions.16,17 In particular, given increasing interests in biologically relevant polymers and environmentally benign synthesis, the straightforward preparation of functional, narrow disperse water-soluble AIE polymers as prepared in aqueous media at low temperatures remains considerably challenging. Noteworthy, difficulties associated with accurate exploration of the structure–property relationship (SPR) of AIE polymers with broad molecular weight distributions emphasize the significance of polymers with low dispersity (Đ).16
Reversible deactivation radical polymerizations (RDRP) and living radical polymerization (LRP), including reversible addition–fragmentation polymerization (RAFT),22,23 atom-transfer radical polymerization (ATRP)24 and Cu(0)-mediated reversible deactivation radical polymerization (Cu(0)-RDRP),25,26 allow for the synthesis of well-defined polymers and (multi)block copolymers with quite sophisticated architectures even in aqueous media.27 Of these techniques, the use of RAFT has been predominant, especially for polymerization of acrylamides in water and aqueous media.28,29 Indeed, acrylamides have often been demonstrated to be problematic to be polymerized by traditional ATRP approaches with less control over molecular weights or limited synthesis of block copolymers. Perrier et al. reported the multiple block copolymerization of comprising up to four different acrylamide monomers with a degree of polymerization (DP) = 10 for each block using RAFT in both aqueous (70 °C) and organic media (65 °C).28 In a separate study, the same group presented the production of decablock poly(N,N-dimethylacrylamide) (PDMA) and homopoly(4-acryloylmorpholine) (PNAM) with a final Đ ∼ 1.15 after 10 block extensions employing the same technique.29 However, a prolonged reaction time (24 h) for each block was required in both cases. Of particular relevance to AIE, the majority RDRP to polymerize acrylamides were limited in organic solvents. Zhu et al. prepared dual-responsive copolymers with AIE features by incorporating a tetraphenylethylene (TPE)-based monomer with poly(N-isopropylacrylamide) (PNIPAM), a well-adopted thermosensitive polymer.30 The copolymerizations were performed in DMF at 70 °C using the RAFT method for more than 24 h and the resultant polymers had a compromised dispersity (Đ ∼ 1.41). Yuan et al. reported the copolymerization of a TPE-containing monomer with benzyl methacrylate using PDMA macro-chain-transfer agents combing RAFT and polymerization-induced self-assembly (PISA) in ethanol, in which the correlation of the AIE effect to the nanostructure of polymer assemblies was explored.31
Despite this elegant process, high temperatures and/or a relatively long time frame are routinely applied in aqueous RAFT polymerisation, potentially limiting their applications in biological science. The Haddleton group introduced a novel synthetic route to perform aqueous copper-mediated RDRP via the in situ formation of Cu containing catalysts exploiting the rapid disproportionation (<10 seconds) as an advantageous driving force.32–34 ATRP in water has often been described as being problematic with a Cu/tris(2-pyridylmethyl)amine (TPMA) complex being described as being the best and most versatile catalyst to employ in water35 even though the quite popular Me6Tren has been shown to be efficient.32–34 This rapid rate of polymerization requires a high radical concentration at any moment in time, however, the excellent control we were seeking is often thought to require a low concentration of radicals, which is somewhat contradictory. Ballard and Asua have described how this apparent contradiction can be explained by taking into account radical diffusion time which explains this rapid and efficient polymerization.36 We decided to use the route we had previously developed using Me6Tren as this proved convenient and effective comforted with the explanation provided by Ballard and Asua. This strategy paves the way for the design and synthesis of an array of hydrophilic polyacrylamides and polyacrylates at sub-ambient temperatures with full monomer conversions producing polymers with narrow dispersities (Đ < 1.10) attained in just few minutes.26,37 In addition, high end-group fidelity is retained even at high conversions, thereby allowing in situ chain extensions and multiblock copolymerizations by iterative monomer addition if required.38 This chemistry was further demonstrated to be compatible with multiple functional groups and biologically associated media such as blood serum39 and PBS.32 Although aqueous Cu-RDRP has been proven to be robust and rapid, there is no report concerning the synthesis of water-soluble polymers with AIE features to date. It is noted that although certain “heavy metals” are often undesirable copper(II) is an essential part of a healthy diet with multiple copper containing enzymes present in the human body and has GRAS (generally regarded as safe) status from the FDA. In addition, copper salts can be easily removed using water swellable complexing resins if required.
Fluorescent organic nanoparticles (NPs) have drawn broad attention in bioluminescence imaging, due to their ready operation, low cost, as well as non-invasive and fast imaging.40 Combining the AIE effect with organic NPs leads to the construction of high-performance fluorescent nanoparticles with excellent biocompatibility and photostability regarding biomedical applications.41–43 It will be even more favourable if the AIE-active NPs could be instinctively generated driven by self-assembly of water-soluble polymers with AIE occurring in water, avoiding often tedious and time-consuming encapsulation of small organic components.
In previous work, the AIEgen-containing initiator tetraphenylethene bromoisobutyrate (TPEBIB) had been synthesized and employed to prepare a range of both hydrophobic and hydrophilic polyacrylates with AIE properties through Cu-wire mediated RDRP in varying organic solvents.44 Herein, we present the synthesis of water-soluble AIE-active polymers with diverse architectures initiated by TPEBIB via aqueous Cu-mediated RDRP at sub-ambient temperatures (Scheme 1).
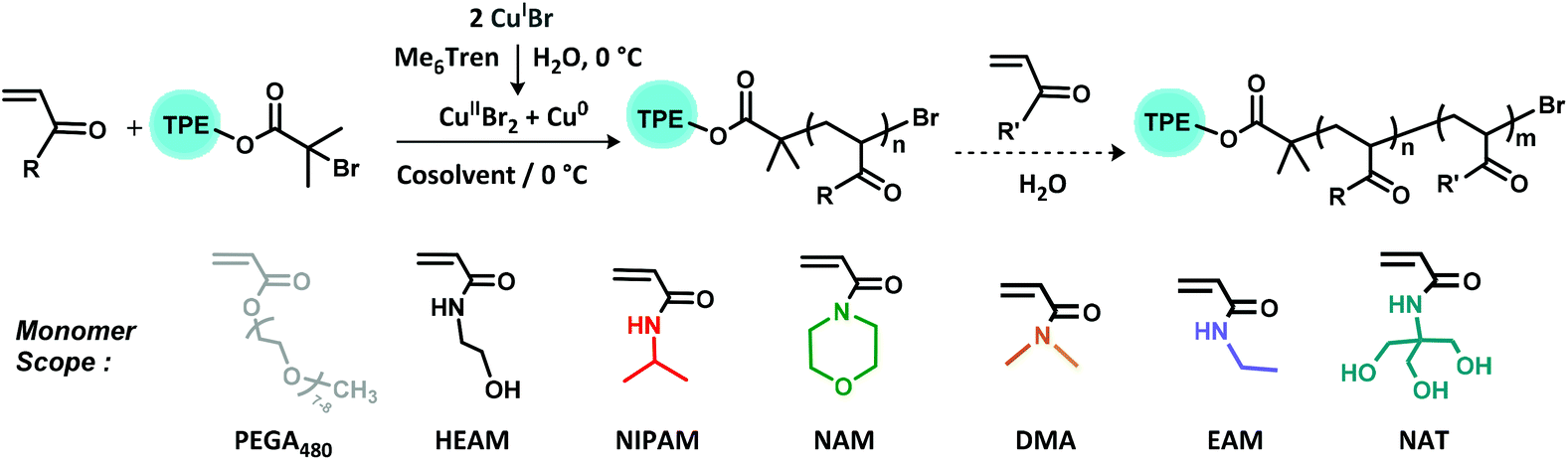 | ||
| Scheme 1 Reaction scheme for homopolymerizations and in situ block copolymerizations of acrylamides and PEGA480 with initiator TPEBIB in aqueous Cu-RDRP at 0 °C. | ||
Results and discussion
Optimization of aqueous Cu-RDRP conditions of HEAM (DP = 100) initiated by TPEBIB
Initially, in order to examine the ability of the hydrophobic TPEBIB to initiate polymerization in aqueous media, aqueous Cu-RDRP of poly((ethylene glycol) methyl ethyl acrylate) (PEGA480) was conducted in a vial with DMSO/water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent, following the conditions developed in a previous report ([I]:[PEGA480]:[Cu(I)Br]:[Me6Tren] = 1:20:0.4:0.4).32 The cosolvent was introduced in order to enhance the solubility of the hydrophobic initiator in the reaction media independently of the disproportionation of copper(I) in pure water as this is a much more rapid reaction than that in mixed water/organic solvents. Thus, it is noted that the catalyst is prepared by in situ disproportionation of Cu(I)Br/Me6TREN in pure water at 0 °C prior to the addition of the monomer/co-solvent mixture is important and a requirement for effective polymerisation (Scheme 1); when organic cosolvents are used the equilibrium constant shifts markedly towards comproportionation as opposed to disproportionation of Cu(I). Pleasingly, the monomer conversion of PEGA480 reached 98% in 20 h, resulting in a final polymer with Mn = 8900 g mol−1 with a dispersity = 1.11 (Fig. S1†). Considering these promising results with TPEBIB as initiator, the analogous polymerization of N-hydroxyethyl acrylamide (HEAM) targeting a moderate DP = 100 was attempted. However, surprisingly only 15% monomer conversion was achieved after 20 h (Table 1, entry 1). Thus, other cosolvents were applied to the binary system (Table 1, entries 2–5). Amongst them, THF and dioxane, with lower polarity of 0.207 and 0.164 respectively, showed much better performance than that of the more polar cosolvents DMSO, DMF and MeCN (0.444, 0.386, and 0.460).45 The cosolvents with lower polarity (THF and dioxane) tended to be better solvents for the solubilisation of TPEBIB. The widely used alcoholic media previously used in aqueous Cu-RDRP,46,47 were not explored in this work due to their limited solubility of TPEBIB in these solvents.
:1) solvent, following the conditions developed in a previous report ([I]:[PEGA480]:[Cu(I)Br]:[Me6Tren] = 1:20:0.4:0.4).32 The cosolvent was introduced in order to enhance the solubility of the hydrophobic initiator in the reaction media independently of the disproportionation of copper(I) in pure water as this is a much more rapid reaction than that in mixed water/organic solvents. Thus, it is noted that the catalyst is prepared by in situ disproportionation of Cu(I)Br/Me6TREN in pure water at 0 °C prior to the addition of the monomer/co-solvent mixture is important and a requirement for effective polymerisation (Scheme 1); when organic cosolvents are used the equilibrium constant shifts markedly towards comproportionation as opposed to disproportionation of Cu(I). Pleasingly, the monomer conversion of PEGA480 reached 98% in 20 h, resulting in a final polymer with Mn = 8900 g mol−1 with a dispersity = 1.11 (Fig. S1†). Considering these promising results with TPEBIB as initiator, the analogous polymerization of N-hydroxyethyl acrylamide (HEAM) targeting a moderate DP = 100 was attempted. However, surprisingly only 15% monomer conversion was achieved after 20 h (Table 1, entry 1). Thus, other cosolvents were applied to the binary system (Table 1, entries 2–5). Amongst them, THF and dioxane, with lower polarity of 0.207 and 0.164 respectively, showed much better performance than that of the more polar cosolvents DMSO, DMF and MeCN (0.444, 0.386, and 0.460).45 The cosolvents with lower polarity (THF and dioxane) tended to be better solvents for the solubilisation of TPEBIB. The widely used alcoholic media previously used in aqueous Cu-RDRP,46,47 were not explored in this work due to their limited solubility of TPEBIB in these solvents.
| Entrya | Cosolventb | Cu(I):L |
Time (h) | Con.c (%) |
M
n,SECd (g mol−1) |
Đ |
|---|---|---|---|---|---|---|
|
a In all reactions, the in situ disproportionation was conducted in 1 ml H2O in an ice bath (0 °C). The total volume of the organic-water solution = 4 ml. [DPn]:[TPEBIB]:[Cu(I)Br]:[Me6Tren] = 100:1:0.8:0.4/0.6/0.8.
b Cosolvent: dimethyl sulfoxide (DMSO), dimethylformamide (DMF), acetonitrile (MeCN), tetrahydrofuran (THF). 3 ml cosolvent was used for entries 1–10. 2 ml dioxane was used for entry 11.
c Conversions were calculated according to 1H NMR in DMSO-d6.
d Determined by SEC employing DMF as eluent calibrated by narrow PMMA molecular weight standards.
e The volume ratio of dioxane to water is 1:1.
|
||||||
| 1 | DMSO | 0.8:0.4 |
20 | 15 | 10200 |
1.24 |
| 2 | DMF | 0.8:0.4 |
20 | 9 | 1600 | 1.08 |
| 3 | MeCN | 0.8:0.4 |
20 | 26 | 6500 | 1.12 |
| 4 | THF | 0.8:0.4 |
20 | 61 | 16700 |
1.12 |
| 5 | Dioxane | 0.8:0.4 |
20 | 51 | 12000 |
1.11 |
| 6 | THF | 0.8:0.6 |
3 | 81 | 21000 |
1.15 |
| 7 | THF | 0.8:0.8 |
3 | 88 | 26500 |
1.21 |
| 8 | Dioxane | 0.8:0.6 |
4 | 83 | 20400 |
1.11 |
| 9 | Dioxane | 0.8:0.8 |
4 | 96 | 23100 |
1.14 |
| 10 | Dioxane | 1.0:1.0 |
3 | 94 | 24700 |
1.15 |
| 11e | Dioxane | 0.8:0.8 |
1 | 99 | 17500 |
1.08 |
Although the monomer conversions increased significantly (>50%) by using THF or dioxane as the cosolvents, it is still not an ideal system due to the long reaction time required to reach high monomer conversion (20 h). In order to increase the polymerization rate, the concentration of the ligand with respect to the Cu(I)Br was increased with THF or dioxane as the cosolvent (Table 1, entries 6–9). Indeed, the increased [Me6TREN] contributes to a higher conversion in a shorter time (Table 1, entries 6, 7 vs. 4; entries 8, 9 vs. 5). When [CuBr]:[Me6TREN] = 0.8:0.8, monomer conversion of 88% (THF/water, 3 h) and 96% (dioxane/water, 4 h) were attained, with slightly broader molecular weight distributions (Đ = 1.21 and 1.14, respectively) (entries 7 and 9). The higher Đ values were attributed to relatively less effective deactivation due to the lower concentration of additional copper(II) halides, nevertheless, these dispersities were seen as being acceptable. An attempt using a higher copper-to-ligand ratio corresponding to the initiator (1.0:1.0) failed to speed the reaction or yield a more controlled polymer (Table 1, entry 10). Taking both the dispersity and polymerization rate into account, dioxane was chosen as the desired cosolvent with a [CuBr]:[Me6TREN] = 0.8:0.8 for further polymerisations. Since the equilibrium constant (the balance between active and dormant species) is considerably higher in water than that in less polar organic solvents,48 the volume ratio of dioxane to water was further reduced from 3:1 to 1:1 (Table 1, entry 11). As expected, the polymerization was significantly faster, attaining 99% monomer conversion in 1 h. Remarkably, the dispersity was as low as 1.08 under these conditions. Thus the conditions of aqueous Cu-RDRP of N-hydroxyethyl acrylamide using hydrophobic TPEBIB initiator were optimized to be [TPEBIB]:[CuBr]:[Me6TREN] = 1:0.8:0.8 in dioxane/water (1:1) binary mixture, which were applied to all the remaining polymerizations (otherwise stated) in this work.
Aqueous homopolymerizations: monomer scope, kinetic analysis, and varying DPs
In addition to HEAM, other commercial hydrophilic monomers, including N-isopropylacrylamide (NIPAM), 4-acryloylmorpholine (NAM), N,N-dimethylacrylamide (DMA), N-ethylacrylamide (EAM) and N-[tris(hydroxymethyl)methyl] acrylamide (NAT) were also successfully polymerized targeting DP = 100 under identical optimized conditions (Fig. 1A–F). Homopolymerizations of these various monomers reached full monomer conversion (≥99%) within 1–2 h. The monomodal mass distribution, narrow dispersities (Đ = 1.08–1.15), as well as the excellent correlation between the theoretical (Mn,th) and the experimental molecular weights (Mn,NMR and Mn,SEC), collectively indicated good controlled/living characteristics of this chemistry. This is in accordance with the explanation put forward by Ballard and Asua.36 | ||
| Fig. 1 DMF-SEC molecular weight distribution of synthesized TPE-terminated polyacrylamides. (A) TPE-PHEAM100, (B) TPE-PNIPAM100, (C) TPE-PNAM100, (D) TPE-PDMA100, (E) TPE-PEAM100, (F) TPE-PNAT100, (G) TPE-PNIPAM with targeted DP = 50–1000, (H) in situ block copolymerization by sequential monomer addition; TPE-PNIPAM90 (light pink) and TPE-PNIPAM90-b-PNAM10 (pink), (I) statistical polymerization of TPE-PNIPAM90-s-PNAM10. | ||
The purified polymers were also characterized by NMR (Fig. S2 and S3†) and FT-IR (Fig. S4†) to investigate the different architectures. The attachment of the TPE group to polymers was exhibited by the signals δ = 7.15–6.77 ppm in 1H NMR spectrum, δ = 150–120 ppm of 13C NMR spectrum, together with the overlapping traces of the RI and UV (λex = 309 nm) in the SEC (Fig. S5†). Additionally, the characteristic peaks of the side chains were assigned (Fig. S2–S4†).
In order to further demonstrate the effectiveness of the system and the polymerization rate, kinetic investigations were conducted for the homopolymerizations of HEAM and NIPAM (Fig. S6† and Fig. 2A, B). The results revealed near-100% monomer conversions in <1 hour (95% in 30 min for HEAM and 98% in 45 min for NIPAM). It is highlighted that ln([M]0/[Mt]) increased linearly with respect to the reaction time in the dioxane/water media, indicating the polymerization rate to be first order in monomer concentration. This is quite remarkable compared with the wholly aqueous system, where achieving linear first-order kinetics proved challenging due to the extremely fast reaction.34,37 Furthermore, a linear dependence of molecular weight with respect to monomer conversion (Fig. 2B) and low dispersity values (Đ ∼ 1.10) during the reaction also verified that the polymerizations proceed in a well-controlled manner. Matrix assisted laser desorption–ionization time-of-flight mass spectrometry (MALDI-ToF-MS) revealed the presence of the TPE end group with a repeating unit of mass 113.04, corresponding to NIPAM (Fig. 2C and D).
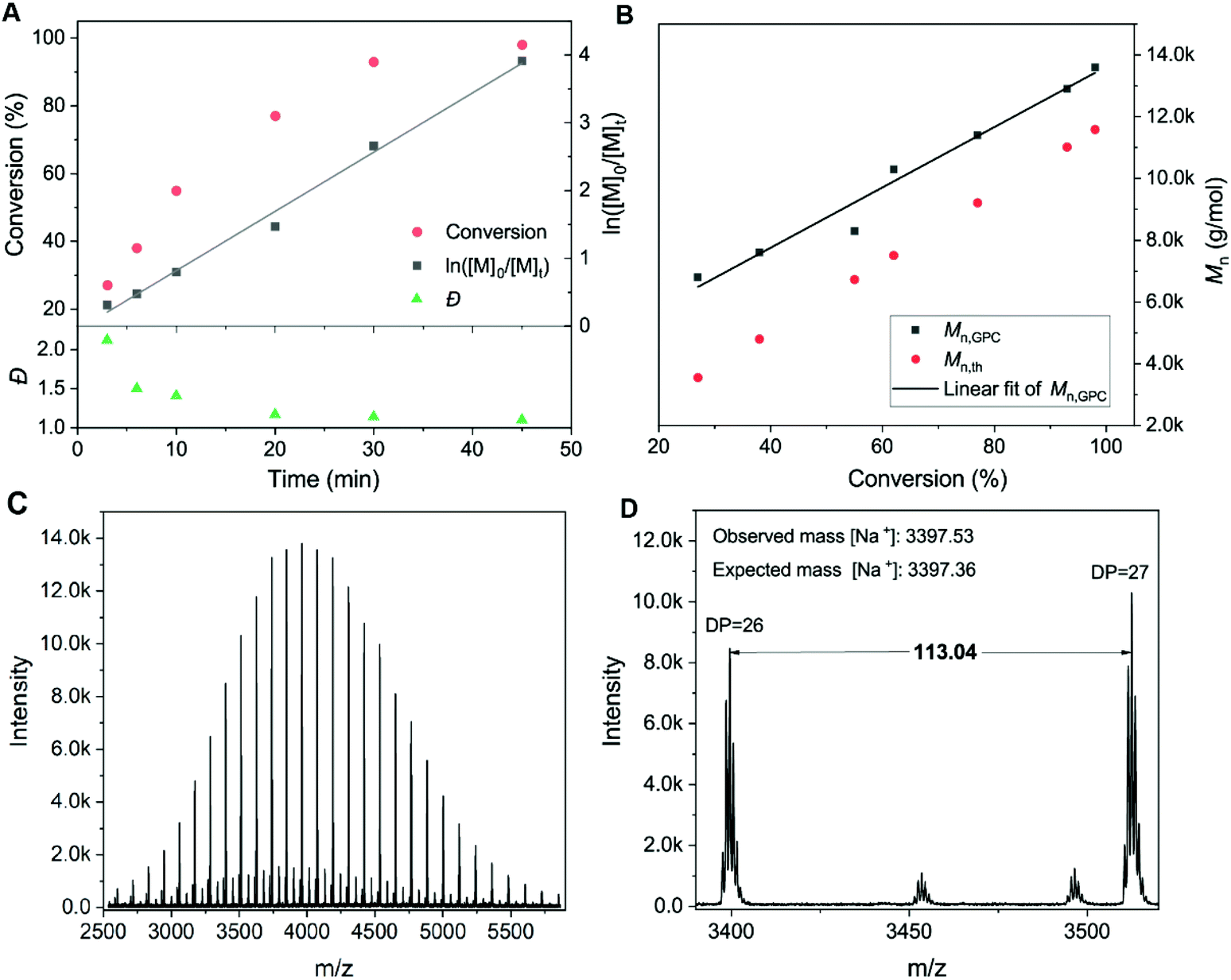 | ||
| Fig. 2 (A) Kinetic study of aqueous Cu-RDRP of TPE-PNIPAM100; (B) comparison of Mn,GPC and theoretical molecular weights. (C and D) MALDI-ToF-MS spectra of TPE-PNIPAM25 synthesized via Cu-RDRP in aqueous solution. | ||
With an increasing interest in thermally responsive polymer assemblies, the homopolymerization of NIPAM targeting differing molecular weights (DP = 50–1000) was attempted to probe the potential of aqueous Cu-RDRP initiated by TPEBIB to give thermoresponsive polymers with AIE properties for use in sensor applications (Fig. 1G and Table S1†). It has been reported that it can be important to optimise the copper-to-ligand ratio when targeting different DPs to afford the most well-defined polymers using aqueous RDRP.32,37,48 Significantly, in this present study, all polymerizations proceeded to near quantitative conversions (≥98%) without further optimizing this reagent ratio, and were well-controlled with a dispersity of 1.09–1.17 within 1 or 2 h even when DP was as high as 1000 (Mn = 114 kDa). Symmetrical monomodal shifts were evidently observed from the SEC traces (Fig. 1G), indicative of excellent control when high molecular weights were targeted.
Block and statistical copolymerizations of PNIPAM and PNAM
In order to obtain AIE polymers with high end group fidelity, in situ chain extensions from PNIPAM were carried out using sequential monomer addition. Copolymerization of PNIPAM and PNAM has been reported to be somewhat problematic with limited conversion of the second PNAM block (56%) due to chain termination events preventing efficient product formation.49 However, we decided to revisit this monomer pair especially as NAM has been less explored using copper-mediated RDRP than that in the case of RAFT. The total DP was fixed at 100 with a change in molar ratio for a series of block copolymers. NIPAM (targeted DP = 50, 70, 80, 90, and 95) was firstly polymerized employing the previous conditions, sampled after 35 min, prior to the second aliquot of deoxygenated NAM in aqueous solution being injected into the reaction vial. Samples were taken again after 40 min. For comparison, the statistical copolymerization experiments were also carried out by adding the mixture of both monomers at the beginning of polymerization.For the block copolymers (PNIPAM-b-PNMA), the conversion of both the first and second block was ≥95% (Table 2). SEC analysis revealed monomodal peaks with a shift of the mass distribution to higher molecular weights upon the second block addition whist maintaining narrow dispersities (Đ ∼ 1.15) (Fig. 1H and Fig. S7†), indicative of full reaction of PNIPAM chains with additional NAM, thus suggesting excellent end group fidelity at high monomer conversions. Of the statistical copolymers (PNIPAM-s-PNMA), total monomer conversion reached ≥98% within 1 or 2 h, yielding polymers with low molecular weight distributions (Đ = 1.11–1.17) (Table 2, Fig. 1I and S8†). In all cases, the negligible deviation between the theoretical (Mn,th) and experimental molecular weights (especially Mn,th) (Table 2) as well as monomodal SEC traces without tailing (Fig. S7 and S8†) was demonstrated. Chemical compositions were characterized by NMR and FT-IR (Fig. S2–S4†).
| Copolymersa | Con.b (%) | M n,th (g mol−1) |
M
n,SECc (g mol−1) |
M n,NMR (g mol−1) | Đ |
|---|---|---|---|---|---|
|
a Copolymerization was conducted using the conditions [TPEBIB]:[CuBr]:[Me6TREN] = 1:0.8:0.8 in dioxane/water (1:1) binary mixture. Expect TPE-PNIPAM50-s-PNAM50 (2 h), the other statistical copolymerizations were stopped in 1 h.
b Conversions were calculated according to 1H NMR in CD3OD; the values in the brackets are the conventions of the first block.
c Determined by SEC employing DMF as eluent calibrated by narrow PMMA molecular weight standards.
|
|||||
| TPE-PNIPAM95-b-PNAM5 | >99 (99) | 12000 |
16000 |
12300 |
1.15 |
| TPE-PNIPAM90-b-PNAM10 | >99 (96) | 12100 |
15900 |
12400 |
1.15 |
| TPE-PNIPAM80-b-PNAM20 | >99 (97) | 12400 |
16500 |
12700 |
1.16 |
| TPE-PNIPAM70-b-PNAM30 | 99 (98) | 12700 |
16900 |
13600 |
1.17 |
| TPE-PNIPAM50-b-PNAM50 | 96 (95) | 13200 |
17400 |
14000 |
1.15 |
| TPE-PNIPAM95-s-PNAM5 | 98 | 12000 |
15700 |
12300 |
1.14 |
| TPE-PNIPAM90-s-PNAM10 | 99 | 12100 |
15700 |
12500 |
1.12 |
| TPE-PNIPAM80-s-PNAM20 | 98 | 12400 |
15400 |
12500 |
1.17 |
| TPE-PNIPAM70-s-PNAM30 | 99 | 12700 |
14500 |
12800 |
1.15 |
| TPE-PNIPAM50-s-PNAM50 | >99 | 13200 |
16500 |
13400 |
1.11 |
In addition, the thermal properties of the obtained polymers were characterized by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements (Fig. S9 and S10†). The hydroxyl-containing polymers, TPE-PHEAM100 and TPE-PNAT100, showed two degradation stages with a decomposition temperature (Td) at 5% weight loss under nitrogen at 292 °C and 253 °C, respectively. The other polymers all had one degradation stage, losing 5 wt% at temperatures ranging between 383 and 409 °C. DSC analysis demonstrated the glass transition temperature (Tg) of these polyacrylamides was in the range of 126–168 °C, of which TPE-PNAM100 had the highest Tg. With the increase of DP from 50 to 1000, the Tg of homopolymer TPE-PNIPAM increased from 140 °C to 149 °C. Moreover, when increasing the ratio of PNAM from 0% to 50%, the Tg of block copolymers and statistical copolymers increased from 143 °C to 146 °C and 153 °C, respectively.
Thermoresponsivity of TPE-PNIPAM containing polymers: LCST, aggregate size and morphology
The thermoresponsive properties of the TPE-terminated PNIPAM homopolymers and copolymers were initially studied using UV-vis and dynamic light scattering (DLS). It was found that the transmission of the block copolymer aqueous solution and the corresponding particle size was dynamically changing due to dynamic self-assembly, as exemplified by TPE-PNIPAM90-b-PNAM10 (Fig. S11†). With an increase in the stabilization time, the transmittance of polymer solution at high temperatures increased from 20% to 80% and smaller aggregates were detected. There was no real change observed after being left for 1 day at ambient temperature. Consequently, to ensure the comparability of the data, all samples were left in water at ambient temperature for 1 day prior to measurements.Herein, the cloud point (Tcp) is defined here by the temperature at which a 10% sharp loss in sample transmittance is detected in the heating cycle. The Tcp of homopolymer TPE-PNIPAM (DP = 50) = 34 °C, which is not affected by the DP from 50 (34.3 °C) to 1000 (34.8 °C) (Fig. S12†). Interestingly, by changing the end group of PNIPAM100 from TPEBIB to ethyl α-bromoisobutyrate (EBiB, less hydrophobic/aggregating), the cloud point increased to 39.0 °C (Fig. S12†). Furthermore, the Tcp of EBiB-terminated PNIPAM has a much larger dependence on the DP, decreasing from 43.6 °C to 35.6 °C with an increase in DP from 50 to 400. Thus, there seems to be a significant contribution of the hydrophobic end-moiety (TPE) in the self-assembly behaviour of these AIE polymers in aqueous media.
In order to construct thermoresponsive particles at physiologically temperatures, a hydrophilic comonomer (NAM) was incorporated by both block and statistical copolymerization. The effects of the different copolymerization methods, as well as the comonomer ratio on the LCST, were evaluated (Fig. 3A and B). In the block copolymers (Fig. 3A), there was a distinctly reduced loss of transmittance with an increasing ratio of NAM at elevated temperatures. The Tcp increased to 35.9 °C and 37.1 °C with 5 mol% and 10 mol% NAM, respectively. It was difficult to define the Tcp when the block incorporation ratio of NAM ≥20 mol%, without rapid loss in transmittance. Conversely, all statistical copolymers showed low transmittance (<10%) upon heating, with a linearly increasing Tcp up to 45.1 °C as a function of the DP values of NAM (Fig. 3B and Fig. S13†). The cooling cycles were also recorded for all the polymers, exhibiting similar hysteresis (∼4 °C lower than Tcp) in full transmittance recovery (Fig. S12 and 13†).
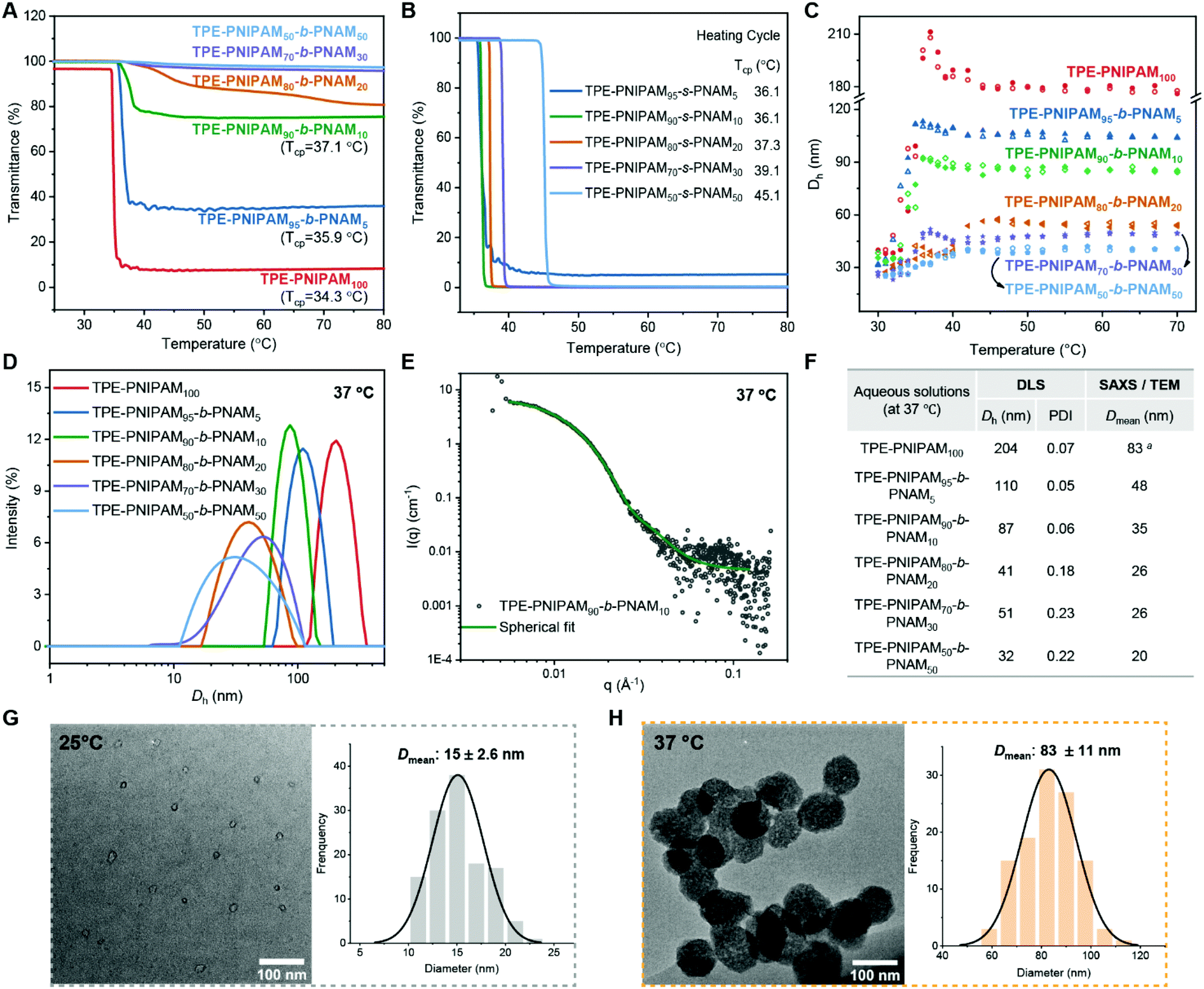 | ||
| Fig. 3 Cloud point curves upon heating of TPE-PNIPAM100 and block copolymers (A) and statistical copolymers (B) with a heating rate of 1 °C min−1. (C) The hydrodynamic diameters of block copolymers as a function of temperature. Solid symbols for heating cycle and hollow symbols for cooling cycle. (D) The particle size distribution by intensity of TPE-PNIPAM100 and block copolymers at 37 °C. (E) Characterization of particle morphology of aqueous solution of TPE-PNIPAM90-b-PNAM10 at 37 °C by SAXS. (F) The comparison of particle sizes determined by DLS and SAXS or TEM. aDetermined by TEM and the others determined by SAXS. The particle morphology and size histograms characterized by TEM at 25 °C (G) and 37 °C (H) of TPE-PNIPAM100. Histograms are from n > 100. All the samples were stabilized in water for 1 day before measurements. [Polymer] = 500 μM for SAXS and [polymer] = 100 μM for the other characterizations. | ||
The particle size information was collected as a function of temperature (30–70 °C) by DLS (Fig. 3C and Fig. S14†). At low temperatures (<LCST), the hydrodynamic diameter (Dh) of all the TPE-PNIPAM containing polymers in aqueous solutions is approximately 30 nm with a polydispersity (PDI) of ∼0.25, due to the self-assembly driven exclusively by the hydrophobic end moiety. When the temperature approaches Tcp upon heating, all the block copolymers tend to self assembles into larger aggregates with a narrower population distribution (Fig. S15†). Increasing the NAM molar ratio from 0 to 50% in the block polymers contributes to decreases in stabilized particle sizes at high temperatures, with Dh varying from 180 nm to 40 nm. By comparison, the reverse trend was found for statistical copolymers (Dh = 240–1600 nm) (Fig. S14†).
Particle behaviour in aqueous solutions at 37 °C were further compared using different techniques (Fig. 3D–H). In terms of TPE-PNIPAM100, TPE-PNIPAM95-b-PNAM5 and TPE-PNIPAM90-b-PNAM10, whose Tcp ≤ 37.1 °C, the Dh values were 204 nm, 110 nm, and 87 nm, respectively (Fig. 3D and F). It is noteworthy distributions (PDI = 0.05–0.07). For the other block copolymers, the Dh = 31–42 nm with a broader dispersity (PDI = 0.18–0.23). Small-angle X-ray scattering (SAXS) was also used (Fig. 3E and Fig. S16†). The mean diameter (Dmean) determined by SAXS is approximately half that obtained from DLS (Fig. 3F). This is as SAXS and transmission electron microscopy (TEM) typically only reflect the internal core or physical diameter, despite which, Dh also includes the thickness of an electrical dipole layer adhered that they all self-assembled to form particles with quite narrow to its surface.50 Additionally, as exemplified by TPE-PNIPAM90-b-PNAM10 (Fig. 3E), all of the particles of TPE-PNIPAM100 and the block copolymers were fitted to be a spherical modal by SAXS (Fig. S16†). TEM was applied with sample preparation at 25 °C (<Tcp) and 37 °C (>Tcp), respectively so as to visualize the changes of the particle size and morphology as a function of temperature. The spherical particles experienced a coil-to-globule transition with a swelling (from 15 nm to 83 nm) due to chain collapse and aggregation of the PNIPAM (Fig. 3G, H and Fig. S17†).
Photophysical properties
As a typical aggregation-induced emission luminogen (AIEgen), TPE shows no emission when fully dissolved but becomes highly luminescent in the aggregated state.51 It was the target that these TPE-terminated polymers would be fluorescent in a wholly aqueous solution. Their UV-vis absorption spectra recorded in water displayed a maximum absorption band at 306–312 nm arisen from the TPE group (Fig. S18†). Subsequently, the photoluminescence (PL) spectra of the homopolymers with different concentrations (0.01–1 mg mL−1) in an aqueous solution at room temperature were measured. The emission intensity was proportional to [polymer] (Fig. 4A, B and Fig. S19†). Taking TPE-PNIPAM100 as an example, distinct emission was observed when the polymer concentration >0.1 mg mL−1 (Fig. 4A). The emissive performance of these 6 homopolymers was compared, among which TPE-PNIPAM100 shows the most intense emission efficiency followed by TPE-PNAM100. Contact angle measurements were used to investigate the structure–property relationship of the spin-coated films of these homopolymers (Fig. 4C). It was found that the water-soluble polyacrylamides, which are more hydrophobic (larger contact angle), were better emitters in aqueous solution, ascribed to aggregation of the TPE groups. The emission of the polymers in media with a variation in water fraction (fw) of THF/water mixtures were measured (Fig. 4D and Fig. S20†). As exemplified by TPE-PNIPAM90-b-PNAM10, the emission was extremely weak at fw < 60%, which was greatly enhanced at fw = 90% and reached the maximum at a 99% water content, 250-fold higher than that in pure THF solution. | ||
| Fig. 4 The PL spectra of TPE-PNAM100 (A) and the peak intensity of the PL spectra (λem = 470 nm) of different TPE-terminated homopolymers (B) in water as a function of concentration at 20 °C. (C) The contact angle of various homopolymers’ film. (D) The PL spectra of TPE-PNIPAM90-b-PNAM10 in water–THF mixtures with different water fractions (fw) measured at 20 °C with [polymer] = 50 μM. The PL spectra of TPE-PNIPAM90-b-PNAM10 (E) and the peak intensity of the PL spectra (λem = 470 nm) of different block copolymers (F) in water as a function of temperature (I is the PL intensity at the corresponding temperature; I0 is emission intensity at 20 °C. [Polymer] = 50 μM). λex = 310 nm for all the measurements. The inserted pictures in (A) and (D) were taken under a UV lamp (λ = 302 nm). | ||
We subsequently investigated how the emission intensity changed as a function of solution temperatures in the range of 20 to 50 °C (Fig. 4E and F). The non-thermoresponsive polymer TPE-PNAM100 was also included for comparison, which had a liner emission decrease trend by gradually elevating the temperature (Fig. 4F). By contrast, in the cases of TPE-PNIPAM-based copolymers, a minor emission decrease was observed between 30 °C and Tcp, attributing to the increasing hydrophobicity on a basis of integral hydrophilicity caused by gradual dehydration of the PNIPAM chains. Moreover, the fluorescence intensity dropped sharply (∼15%) from Tcp to Tcp +1 for TPE-PNIPAM100 and TPE-PNIPAM95-b-PNAM5 aqueous solutions, which is indistinct for the other block copolymers (Fig. 4F). The difference results from the fact that it is more difficult for the UV excitation beam to reach TPE moiety buried in the larger micelles after the coil-to-globule transmission.52 Statistical copolymers also behaved in a similar fashion (Fig. S21†).
Cell imaging
Based on these results, TPE-PNIPAM90-b-PNAM10 with a suitable nanoparticle size in water at physiological temperature (Dh = 87 nm and Dmean = 35 nm measured by DLS and SAXS, respectively) as well as excellent aggregate-state fluorescent was selected to explore the application in cell imaging. The cytotoxicity of TPE-PNIPAM90-b-PNAM10 was first evaluated by a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay using 4T1 cells as the model (Fig. 5A). The cell viability retained >95% at a polymer concentration of up to 30 μg mL−1, indicating excellent biocompatibility of TPE-PNIPAM90-b-PNAM10 with living cells. Subsequently, the cell staining capability of TPE-PNIPAM90-b-PNAM10 was assessed using a confocal laser scanning microscope (CLSM) (Fig. 5B). Bright blue fluorescence within cells was observed after incubation with 10 μg mL−1 polymer for 3 h. The co-localization experiment was further conducted upon co-staining 4T1 cells with LysoTracker Red (LTR), a commercial lysosome imaging dye. The results showed that the staining regions of LTR overlapped well with those of TPE-PNIPAM90-b-PNAM10. The Pearson's correlation coefficient was calculated to be 0.85 (Fig. S22†), suggesting the good lysosome-targeting capability of TPE-PNIPAM90-b-PNAM10. Furthermore, the photostability of the polymer was quantitatively compared with LTR (Fig. 5C). After 60 scans with continuous irradiation (λex = 405 nm), the emission intensity of TPE-PNIPAM90-b-PNAM10 retained over 70% of its initial value, whereas LTR suffered a significant signal loss with only 20% remaining intensity, strongly indicative of the superior photostability of TPE-PNIPAM90-b-PNAM10 to commercial dyes. Evidently, with its low cytotoxicity, high specificity and good photo-bleaching resistance, TPE-PNIPAM90-b-PNAM10 is a promising candidate functioning as lysosome-specific fluorescent probes for bioimaging.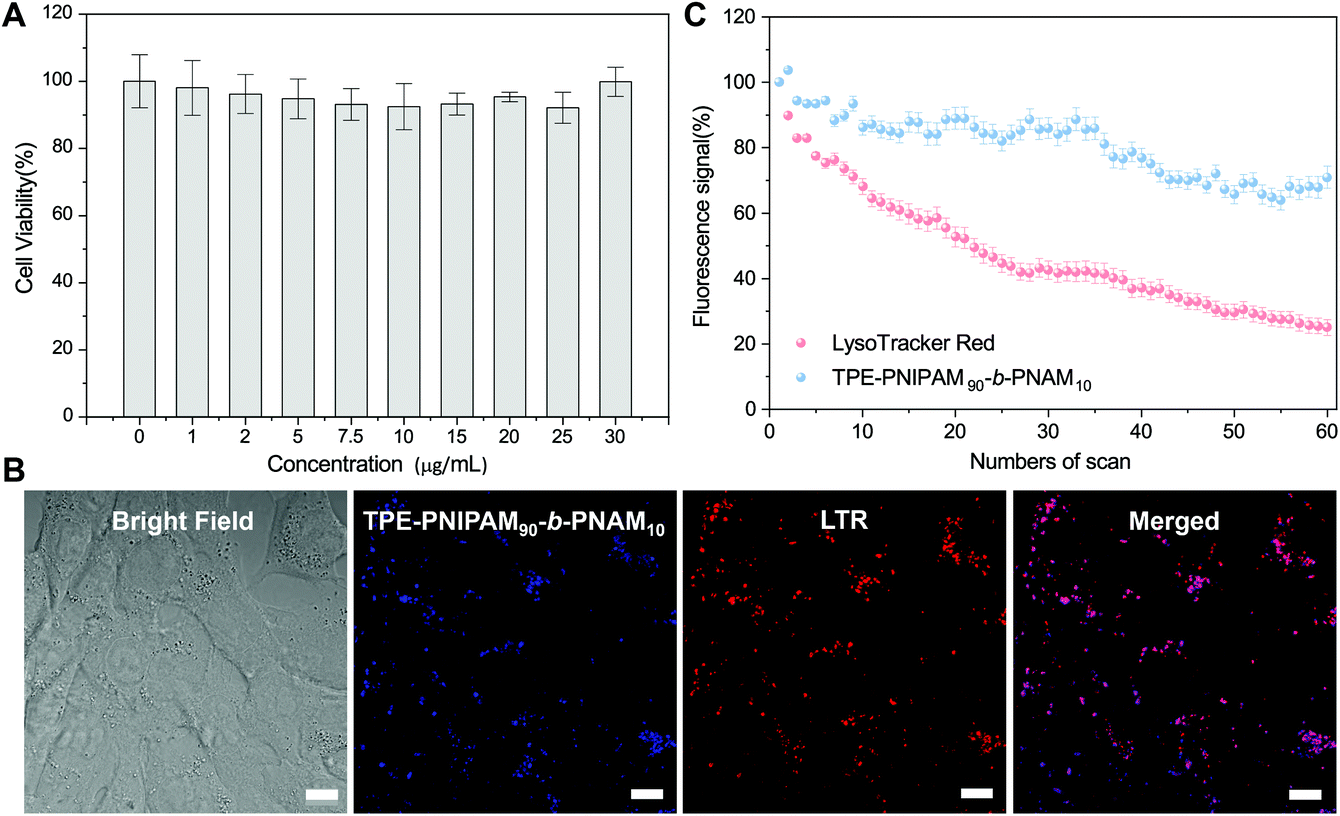 | ||
| Fig. 5 (A) The cytotoxicity of TPE-PNIPAM90-b-PNAM10 to 4T1 cells incubated with different polymer concentrations. (B) Bright field and confocal images of 4T1 cells stained with TPE-PNIPAM90-b-PNAM10 (10 μg mL−1, 3 h), LysoTracker Red (LTR, 1 μM, 30 min) and the merged image. λex: 405 nm (polymer) and 561 nm (LTR); emission filter: 400–600 nm (polymer) and 620–700 nm (LTR). Scale bar = 10 μm. (C) Loss in fluorescence of 4T1 cells stained respectively with TPE-PNIPAM90-b-PNAM10 and LTR as a function of the numbers of the scan of laser irradiation. | ||
Conclusions
In summary, various acrylamides, including NIPAM, were polymerized within 1 or 2 h in a near quantitative fashion, with excellent control over chain lengths (DP = 50–1000) and molecular weight distributions (Đ = 1.08–1.17). The polymerisations show good linear first order kinetics and successful in situ block copolymerizations are possible. A library of thermoresponsive block and statistical copolymers with TPE terminus were obtained, which were demonstrated to possess varying lower critical solution temperature (LCST) and particle sizes depending on the compositions and copolymerization methods. Moreover, all the TPE-labelled polymers were shown to be AIE-active and the photo-physical properties exhibited dependence on the polymer concentration, side chain, as well as temperature. TPE-PNIPAM90-b-PNAM10, spontaneously self-assembled into fluorescent NPs with a suitable size at physiological conditions, was exemplified to be applicable for lysosome-specific cell imaging owing to its low cytotoxicity, high specificity and good photostability. By changing the compositions and copolymerization method of the TPE-PNIPAM-based polymers, fluorescent thermoresponsive nanoparticles with tuneable sizes were straightforwardly formed by self-assembly in aqueous media without any encapsulation. The emission intensity was regulated by the hydrophobicity of the polymers as well as their concentration and the temperature of an aqueous solution. Significantly, the PNIPAM-containing polymers possessed better fluorescent performance and resisted the thermo-induced emission reduction. The good biocompatibility and photostability of these emissive nanoparticles were exemplified by TPE-PNIPAM90-b-PNAM10, indicating their potential for lysosome-specific bioimaging application.Author contributions
CM and DH designed the experiments, CM conducted all of the synthesis and most of the characterizations and wrote the draft manuscript. TH and NN conducted the cell imaging experiments. LA conducted the MALDI measurements and helped with data analysis. SE helped the discussions and purification of polymers. DL offered the instrument trainings and expertise, especially with regard to SEC analysis. SH performed the SAXS measurements and analysis. DH supervised the project and made corrections of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from University of Warwick and China Scholarship Council. We are grateful for the Research Technology Platforms (RTP) in University of Warwick, especially Daniel Lester, James Town and Ivan Prokes for providing NMR, SEC, FT-IR, UV-Vis, DSC, DLS, TGA, SAXS, TEM training and equipment. We also acknowledge the collaboration which was funded by the National Natural Science Foundation of China (21905176) and the Science and Technology Plan of Shenzhen (JCYJ20190808142403590).Notes and references
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741, 10.1039/B105159H
.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC
- W. W. H. Lee, Z. Zhao, Y. Cai, Z. Xu, Y. Yu, Y. Xiong, R. T. K. Kwok, Y. Chen, N. L. C. Leung, D. Ma, J. W. Y. Lam, A. Qin and B. Z. Tang, Chem. Sci., 2018, 9, 6118–6125 RSC
- W. Qin, Z. Yang, Y. Jiang, J. W. Y. Lam, G. Liang, H. S. Kwok and B. Z. Tang, Chem. Mater., 2015, 27, 3892–3901 CrossRef CAS
- M. Gao and B. Z. Tang, ACS Sens., 2017, 2, 1382–1399 CrossRef CAS PubMed
- Y.-l. Liu, Z.-k. Wang, W. Qin, Q.-l. Hu and B. Z. Tang, Chin. J. Polym. Sci., 2017, 35, 365–371 CrossRef CAS
- T. Lin, X. Su, K. Wang, M. Li, H. Guo, L. Liu, B. Zou, Y.-M. Zhang, Y. Liu and S. X.-A. Zhang, Mater. Chem. Front., 2019, 3, 1052–1061 RSC
- J.-S. Ni, P. Zhang, T. Jiang, Y. Chen, H. Su, D. Wang, Z.-Q. Yu, R. T. K. Kwok, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2018, 30, 1805220 CrossRef PubMed
- D. Mao, W. Wu, S. Ji, C. Chen, F. Hu, D. Kong, D. Ding and B. Liu, Chem, 2017, 3, 991–1007 CAS
- G. Qi, F. Hu, L. Shi, M. Wu and B. Liu, Angew. Chem., 2019, 131, 16375–16381 CrossRef
- Y. Wang, W. Wu, J. Liu, P. N. Manghnani, F. Hu, D. Ma, C. Teh, B. Wang and B. Liu, ACS Nano, 2019, 13, 6879–6890 CrossRef CAS
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS PubMed
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- T. Han, Z. Yao, Z. Qiu, Z. Zhao, K. Wu, J. Wang, A. W. Poon, J. W. Y. Lam and B. Z. Tang, Nat. Commun., 2019, 10, 5483 CrossRef CAS PubMed
- T. Han, H. Q. Deng, Z. J. Qiu, Z. Zhao, H. K. Zhang, H. Zou, N. L. C. Leung, G. G. Shan, M. R. J. Elsegood, J. W. Y. Lam and B. Tang, J. Am. Chem. Soc., 2018, 140, 5588–5598 CrossRef CAS PubMed
- R. Hu, A. Qin and B. Z. Tang, Prog. Polym. Sci., 2020, 100, 101176 CrossRef CAS
- Z. Qiu, X. Liu, J. W. Lam and B. Z. Tang, Macromol. Rapid Commun., 2019, 40, 1800568 CrossRef PubMed
- H. Ma, C. Qi, C. Cheng, Z. Yang, H. Cao, Z. Yang, J. Tong, X. Yao and Z. Lei, ACS Appl. Mater. Interfaces, 2016, 8, 8341–8348 CrossRef CAS
- R. Yang, Y. Wang, W. Luo, Y. Jin, Z. Zhang, C. Wu and N. Hadjichristidis, Macromolecules, 2019, 52, 8793–8802 CrossRef CAS
- Y. Liu, J. W. Y. Lam, X. Zheng, Q. Peng, R. T. K. Kwok, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Macromolecules, 2016, 49, 5817–5830 CrossRef CAS
- Y. B. Hu, J. W. Y. Lam and B. Z. Tang, Chin. J. Polym. Sci., 2019, 37, 289–301 CrossRef CAS
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS
- T. G. McKenzie, E. Colombo, Q. Fu, M. Ashokkumar and G. G. Qiao, Angew. Chem., Int. Ed., 2017, 56, 12302–12306 CrossRef CAS PubMed
- F. Lorandi, M. Fantin, Y. Wang, A. A. Isse, A. Gennaro and K. Matyjaszewski, ACS Macro Lett., 2020, 9, 693–699 CrossRef CAS
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2015, 116, 835–877 CrossRef
- G. R. Jones, A. Anastasaki, R. Whitfield, N. Engelis, E. Liarou and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 10468–10482 CrossRef CAS
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 639–649 CrossRef CAS
- Y. Zhao, Y. Wu, S. Chen, H. Deng and X. Zhu, Macromolecules, 2018, 51, 5234–5244 CrossRef CAS
- M. Huo, Q. Ye, H. Che, X. Wang, Y. Wei and J. Yuan, Macromolecules, 2017, 50, 1126–1133 CrossRef CAS
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed
- S. Efstathiou, A. M. Wemyss, G. Patias, L. Al-Shok, M. Grypioti, D. Coursari, C. Ma, C. J. Atkins, A. Shegiwal, C. Wan and D. M. Haddleton, J. Mater. Chem. B, 2021, 9, 809–823 RSC
- E. Liarou, Y. Han, A. M. Sanchez, M. Walker and D. M. Haddleton, Chem. Sci., 2020, 11, 5257–5266 RSC
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS
- N. Ballard and J. M. Asua, ACS Macro Lett., 2020, 9, 190–196 CrossRef CAS
- G. R. Jones, Z. Li, A. Anastasaki, D. J. Lloyd, P. Wilson, Q. Zhang and D. M. Haddleton, Macromolecules, 2016, 49, 483–489 CrossRef CAS
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406–417 RSC
- Q. Zhang, Z. Li, P. Wilson and D. M. Haddleton, Chem. Commun., 2013, 49, 6608–6610 RSC
- J.-S. Ni, Y. Li, W. Yue, B. Liu and K. Li, Theranostics, 2020, 10, 1923–1947 CrossRef CAS PubMed
- W. Che, L. Zhang, Y. Li, D. Zhu, Z. Xie, G. Li, P. Zhang, Z. Su, C. Dou and B. Z. Tang, Anal. Chem., 2019, 91, 3467–3474 CrossRef CAS PubMed
- Z. Zhao, C. Chen, W. Wu, F. Wang, L. Du, X. Zhang, Y. Xiong, X. He, Y. Cai and R. T. Kwok, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed
- R. Zhang, G. Niu, Q. Lu, X. Huang, J. H. C. Chau, R. T. K. Kwok, X. Yu, M.-H. Li, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2020, 11, 7676–7684 RSC
- C. Ma, T. Han, M. Kang, E. Liarou, A. M. Wemyss, S. Efstathiou, B. Z. Tang and D. Haddleton, ACS Macro Lett., 2020, 9, 769–775 CrossRef CAS
-
C. Reichardt and T. Welton, Solvents and solvent effects in organic chemistry, John Wiley & Sons, 2011 Search PubMed
- C. Waldron, Q. Zhang, Z. Li, V. Nikolaou, G. Nurumbetov, J. Godfrey, R. McHale, G. Yilmaz, R. K. Randev, M. Girault, K. McEwan, D. M. Haddleton, M. Droesbeke, A. J. Haddleton, P. Wilson, A. Simula, J. Collins, D. J. Lloyd, J. A. Burns, C. Summers, C. Houben, A. Anastasaki, M. Li, C. R. Becer, J. K. Kiviaho and N. Risangud, Polym. Chem., 2014, 5, 57–61 RSC
- Q. Zhang, P. Wilson, A. Anastasaki, R. McHale and D. M. Haddleton, ACS Macro Lett., 2014, 3, 491–495 CrossRef CAS
- G. R. Jones, R. Whitfield, A. Anastasaki and D. M. Haddleton, J. Am. Chem. Soc., 2016, 138, 7346–7352 CrossRef CAS PubMed
- A. Anastasaki, A. J. Haddleton, Q. Zhang, A. Simula, M. Droesbeke, P. Wilson and D. M. Haddleton, Macromol. Rapid Commun., 2014, 35, 965–970 CrossRef CAS PubMed
- C. M. Maguire, M. Rösslein, P. Wick and A. Prina-Mello, Sci. Technol. Adv. Mater., 2018, 19, 732–745 CrossRef CAS PubMed
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353, 10.1039/B904665H
- L. Tang, J. K. Jin, A. Qin, W. Z. Yuan, Y. Mao, J. Mei, J. Z. Sun and B. Z. Tang, Chem. Commun., 2009, 4974–4976, 10.1039/B907382E
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01432c |
| This journal is © The Royal Society of Chemistry 2022 |