
Modulating the spectroscopy and dynamics of a proton-transfer dye by functionalizing with phenyl groups†
Mario
Gutiérrez‡
 a,
Eduardo
García‡
a,
Cristina
Monterde
b,
Félix
Sánchez
b and
Abderrazzak
Douhal
*a
a,
Eduardo
García‡
a,
Cristina
Monterde
b,
Félix
Sánchez
b and
Abderrazzak
Douhal
*a
aDepartamento de Química Física, Facultad de Ciencias Ambientales y Bioquímica, and INAMOL, Universidad de Castilla-La Mancha, Avenida Carlos III, S. N., 45071 Toledo, Spain. E-mail: abdderrazzak.douhal@uclm.es
bInstituto de Química Orgánica General, IQOG-CSIC, Juan de la Cierva, 3, 28006 Madrid, Spain
First published on 21st February 2022
Abstract
Molecules undergoing excited-state proton transfer (ESPT) reactions are among the most interesting systems from spectroscopic and photophysical viewpoints. These molecules can be further functionalized with electron donating or accepting groups, inducing intramolecular charge transfer (ICT) events, which might be coupled to the ESIPT ones, conferring them with different spectroscopic and photophysical properties, which can be essential to implement the related materials in many key scientific and technological fields. Here, we report new benzimidazole derivatives that are functionalized with a phenyl group, 2-(5,10-diphenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenol (DP-HPPI), and its methylated equivalent, 2-(2-methoxyphenyl)-5,10-diphenyl-1H-phenanthro[9,10-d]imidazole (DP-MPPI). The results prove that these molecules in solutions undergo an ultrafast ICT (400–700 fs) reaction. Additionally, DP-HPPI also undergoes a reversible ESIPT process in dichloromethane. However, this is precluded in acetonitrile due to the involvement of intermolecular H-bonds in this solvent. These results provide key insights into the development of proton-transfer materials with bespoke spectral and photodynamical properties.
Introduction
Molecules undergoing excited-state intramolecular charge transfer (ICT) and intramolecular proton transfer (ESIPT) processes are among the most interesting systems, not only from spectroscopic and photodynamical viewpoints,1–13 but also owing to their great potential for employment in advanced technologies such as proton transfer lasers,14,15 anticounterfeiting,16–18 fluorescence sensing,17,19,20 fluorescent probes in biological systems,21,22 or organic light-emitting diodes.23–25 Indeed, over the last four decades, research studies have dedicated great efforts to design, develop, and multidisciplinary explore (theoretically, experimentally and their practical real-world applicability) a vast number of molecules and systems undergoing ICT and ESIPT reactions.26–31 Three molecules stand out among all of them, 2-(2-hydroxyphenyl)benzoxazole (HBO), 2-(2-hydroxyphenyl)benzimidazole (HBI) and 2-(2-hydroxyphenyl)benzothiazole (HBT), as they are the starting point for the generation of many derivatives, showing outstanding optical and photodynamical properties.32–42Interestingly, some of these systems can undergo either ESIPT or ICT, but there exist others where both reactions can be coupled.43–46 The rate constants of ESIPT and ICT strongly depend on the chemical structure and the surrounding environment. Subsequently, some examples have been found where the ESIPT event occurs in shorter times than the ICT one and vice versa. For instance, HBI undergoes an ESIPT process, leading to the formation of keto-type species, which then undergo an ICT reaction favoured by molecular twisting.45,46 Similarly, the ultrafast excited-state photodynamics of a HBO derivative, HBOCE, has been found to exhibit an ultrafast ESIPT (250 fs–1.2 ps) reaction followed by a relatively slower ICT (2.7 ps) reaction.44 On the other hand, the processes can be coupled upside down, being the ICT mechanism faster than the ESIPT one. For example, our group has recently reported that another HBO derivative, 2-(2′-hydroxyphenyl)benzoxazole (5A-HBO), undergoes an ultrafast ICT process (<50 fs) followed by a reversible ESIPT reaction (τDPT = 1.5 ns and τRPT = 1.6 ns) in dichloromethane (DCM), while, in a more basic solvent such as acetonitrile (ACN), the ESIPT process is blocked owing to H-bond interactions with the solvent, and therefore only the ultrafast ICT process was observed.43
Few years ago, we have also reported on the photobehavior of a HBI derivative, 2-(2-hydroxyphenyl)-1H-phenanthro[9,10-d]imidazole (HPPI), which undergoes two distinct ESIPT reactions owing to the presence of different conformations: one faster (2 ps) related to the enol–keto phototautomerism of planar enol conformers and another slower one (12 ps) due to non-planar enol structures.47 Then, the question that arises is whether it is possible to induce an ICT reaction coupled to ESIPT by functionalizing the phenanthrene moiety of the HPPI molecule with electron-rich phenyl groups.
Hence, we have synthesized a new PT-imidazole derivative, 2-(5,10-diphenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenol (DP-HPPI) and its methylated equivalent 2-(2-methoxyphenyl)-5,10-diphenyl-1H-phenanthro[9,10-d]imidazole (DP-MPPI) (Scheme 1), and spectroscopically and photophysically characterized them in two different solvents using a combination of UV-vis steady-state and laser-based time-resolved techniques. The photoexcitation of the DP-MPPI molecule (which cannot undergo an ESIPT event) in the DCM solution induces an ultrafast ICT reaction occurring in 400 fs, which generates two different species having a charge-transfer (CT) character. Similarly, the photoexcited DP-HPPI in DCM undergoes an ultrafast ICT reaction in ∼700 fs, leading to the formation of two different conformers, one of which undergoes a subsequent reversible ESIPT process generating the keto (K) tautomer. Interestingly, when DP-HPPI is photoirradiated in the ACN solution, the ESIPT reaction is inhibited, most probably because of the establishment of an intermolecular H-bond with ACN molecules. In this case, we only observed the emission and formation of the CT species. The results presented herein highlight the possibilities of tuning the optical and photophysical properties of ESIPT dyes by inducing an ICT reaction through chemical engineering, providing new insight into the design of these types of molecules.
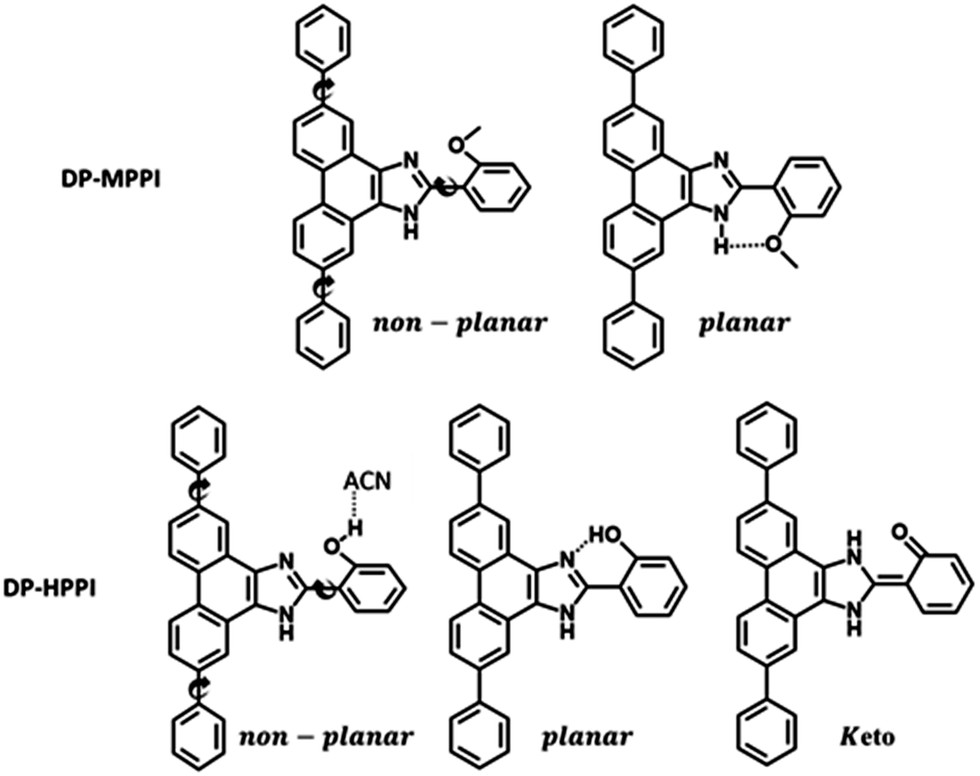 | ||
| Scheme 1 Proposed molecular structures of DP-MPPI and DP-HPPI molecules discussed in this work. | ||
Experimental section
The synthesis, purification and characterization of 2-(5,10-diphenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenol (DP-HPPI) and its methylated equivalent 2-(2-methoxyphenyl)-5,10-diphenyl-1H-phenanthro[9,10-d]imidazole (DP-MPPI) (Scheme 1) are described in the ESI,† together with the 1H NMR (in DMSO-d6) and FTIR (in KBr) spectra of these molecules (Fig. S1 and S2, ESI,† respectively).The used anhydrous solvents (dichloromethane (99.9%, DCM) and acetonitrile (99.8%, ACN) were purchased from Scharlab and Sigma-Aldrich, respectively, and were used as received.
The steady-state UV-visible absorption and fluorescence spectra were recorded using JASCO V-670 and FluoroMax-4 (Jobin-Yvon) spectrophotometers, respectively.
Picosecond (ps) emission decays were recorded using a ps-time-correlated single photon counting (TCSPC) system.48 Upon excitation at 371 nm, the sample was excited using a 40 ps pulsed diode laser (<5 mW, 40 MHz). The emission signal was collected at a magic angle and the instrument response function (IRF) was ∼70 ps. The IRF of the system was measured using a standard LUDOX solution (Sigma-Aldrich). The decays were deconvoluted and fitted to a single or multiexponential function using the FLUOFIT package (PicoQuant), allowing single and global fits. The quality of the fit was estimated by χ2(which was always below 1.2) and the distribution of the residues.
The femtosecond (fs) emission transient decays were collected using the fluorescence up-conversion technique. The system consists of a fs Ti:sapphire oscillator MaiTai HP (Spectra Physics) coupled to second harmonic generation and up-conversion setups.49 The oscillator pulses (90 fs, 250 mW, 80 MHz) were centred at 640 nm and doubled in an optical setup through a 0.5 mm BBO crystal to generate a pumping beam at 320 nm (∼0.1 nJ per pulse). The polarization of the latter was set to the magic angle with respect to the fundamental beam. The sample was placed in a 1 mm thick rotating cell. The fluorescence was focused with reflective optics into a 1 mm BBO crystal and gated with the fundamental fs-beam. The IRF of the full setup (measured as a Raman signal of the pure solvent) was ∼250 fs. To analyse the decays, the multiexponential function convoluted with the IRF was used to fit the experimental transients. Using this procedure, we can resolve components of ∼50 fs. All the experiments were performed at 293 K.
Results and discussion
Steady-state UV-visible absorption and emission studies
To begin with, we have explored the steady-state optical properties of DP-MPPI and DP-HPPI dissolved in DCM and ACN solutions. Fig. 1A and B show the UV-visible absorption and emission spectra of both molecules in DCM and ACN solutions, respectively. DP-MPPI and DP-HPPI in the DCM solution show very similar absorption spectra with intensity maxima at 292, 321 and 376 nm (εDP-MPPI(376nm) = 1.3 ± 0.2 × 104 M−1 cm−1 and εDP-HPPI(376nm) = 1.1 ± 0.2 × 104 M−1 cm−1), which are assigned to the S0 (π) → S2 (π*) and S0 (π) → S1 (π*) transitions of enol tautomers, respectively. Compared to those of the unfunctionalized molecules (MPPI and HPPI in DCM),47 the absorption spectra are shifted to longer wavelengths (by ∼16 nm, 1180 cm−1) and have less vibrational structures. This reflects the influence of phenyl terminal groups, which increase the π-electronic density of the molecule and may induce an ICT reaction in the excited molecules.47 Moreover, the phenyl groups can rotate or twist around their axis, causing a loss of the well-resolved vibrational structures (Scheme 1). The absorption spectra of both molecules in ACN are very similar and also comparable to the ones observed in DCM, indicating that the solvent properties do not affect the ground state (S0) species of DP-MPPI and DP-HPPI (Fig. 1B).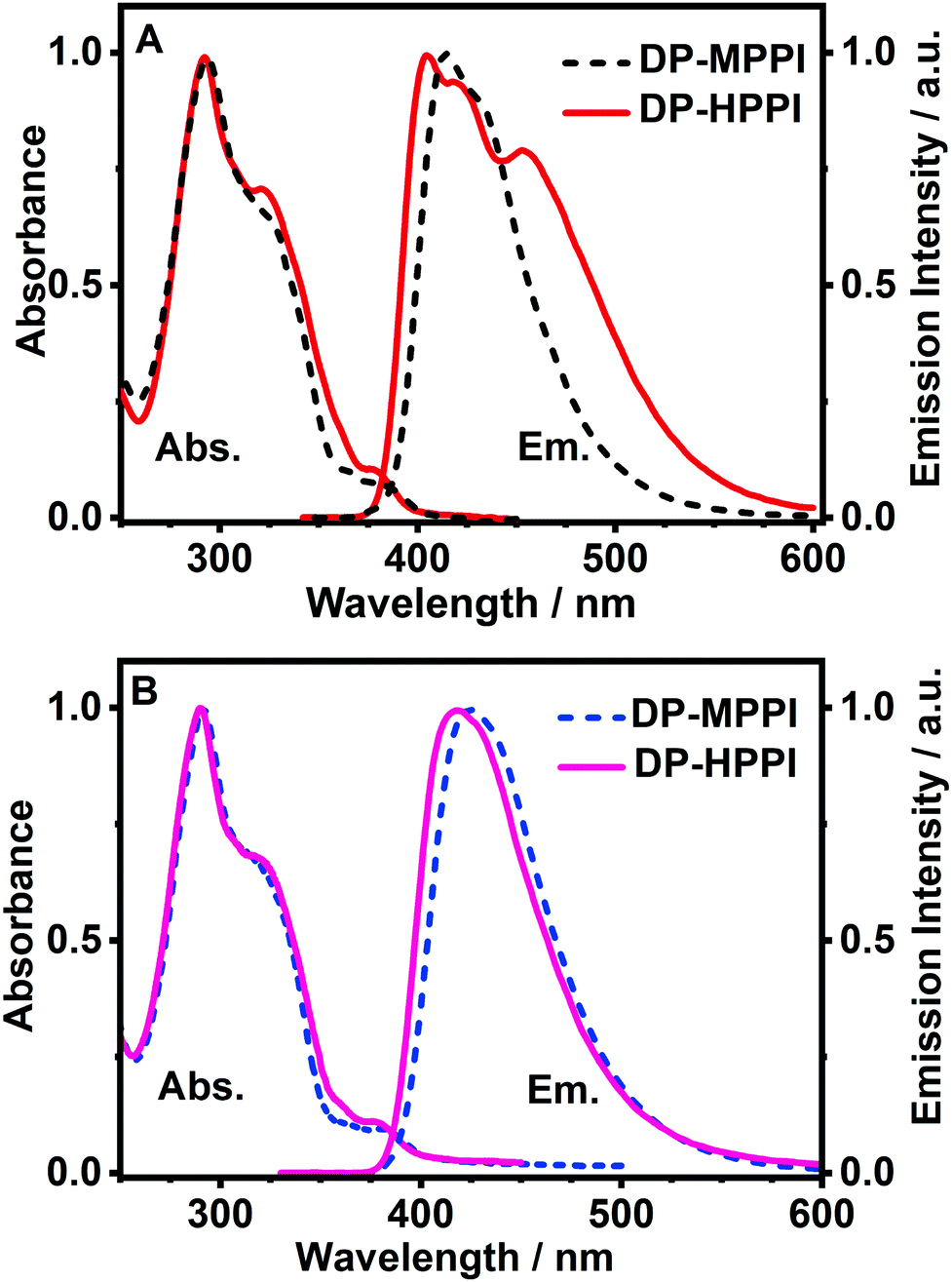 | ||
| Fig. 1 Normalized UV-visible absorption and emission spectra of DP-MPPI and DP-HPPI in (A) DCM and (B) ACN solutions. For emission spectra, the excitation wavelength was 320 nm. | ||
In contrast, the fluorescence spectra of these molecules in the DCM solutions show spectral differences, such as the presence of an additional red-shifted emission band and a shift to shorter wavelengths of the main emission band for the DP-HPPI molecule (Fig. 1A). The emission spectrum of DP-MPPI in DCM consists of a band with an intensity maximum at 415 and a peak at 430 nm, independently of the excitation wavelength. Compared to the unfunctionalized molecule (MPPI) whose Stokes shift is ∼1100 cm−1, the large Stokes shift (more than 2 times higher, ∼2500 cm−1) observed for DP-MPPI suggests that the ICT reaction occurs at S1. This process may take place from the diphenyl groups, which has a high electronic density, to the methoxyphenyl moiety. Indeed, a combination of TDDFT calculations and experimental evidences obtained for the parent protonated molecule (HPPI) indicates the charge redistribution from the phenanthrene part to the phenol one,47 and, therefore, this ICT will be enhanced by the presence of the electron-rich phenyl groups. Moreover, similar ICT reactions have been reported for other ESIPT molecules functionalized with electron-donating substituents, reinforcing our assumption.43,50–52 For example, the 6-amino-2-(2-methoxyphenyl)benzoxazole (6A-MBO) molecule exhibited an Stokes shift of 7190 cm−1 due to the ICT process occurring from the electron-donating amino group to the methoxy-phenyl part.50
The emission spectrum of DP-HPPI in DCM is more complex than its methylated derivative (Fig. 1A). It is structured and displays a band with an intensity maximum at 405 nm and a peak at 420 nm; however, an additional red-shifted band with an intensity maximum at 453 nm is also detected. Similar to DP-MPPI, the Stokes shift considering the blue shifted band (405 nm) is large (∼1905 cm−1), reflecting a photoexcited ICT reaction with the subsequent emission of the species having a CT character. On the other hand, the red-shifted fluorescence band is assigned to the emission of the K tautomer formed as a result of the ESIPT reaction in the excited enol charge transfer (ECT) species.
In order to shed more light on the luminescence properties of these molecules, we have also recorded the emission spectra of DP-MPPI and DP-HPPI in ACN, which is a more polar and basic solvent (f(ε, n)ACN = 0.71 and β = 0.40) than DCM (f(ε, n)DCM = 0.47 and β = 0.1). The emission spectra of these molecules in ACN are different from those observed in DCM. For instance, the spectra of DP-MPPI and DP-HPPI display a single emission band with their intensity maxima centered at 426 and 420 nm, respectively. The large Stokes shift (∼3120 cm−1) compared to the unfunctionalized molecule (MPPI, ∼1110 cm−1) also indicates the occurrence of the ICT process with the subsequent formation of CT species emitting at longer wavelengths, which is similar to what we observed in DCM. However, compared to the emission spectra obtained in DCM solutions, this band is slightly red shifted, being a sign of the stabilization of the CT species in ACN (more polar than DCM). Interestingly, the emission spectrum of DP-HPPI in the ACN solution does not show the red-shifted emission band observed in DCM and is attributed to the emission of the K tautomer, indicating that it does not undergo the ESIPT reaction in this solvent. The H-bond character of ACN due to its carbonitrile moiety favors intermolecular H-bond interactions with the OH group of DP-HPPI, precluding the occurrence of the ESIPT reaction in the open enol form (Scheme 1). Similar results were reported for the molecule 5-amino-2-(2′-hydroxyphenyl)benzoxazole (5A-HBO).43 This molecule has undergone an ultrafast ICT reaction followed by an ESIPT reaction in n-heptane and DCM solutions. However, in the presence of more basic solvents, such as ACN, tetrahydrofuran (THF), and N,N-dimethylformamide (DMF), the ESIPT reaction was inhibited due to the intermolecular H-bonds between the –OH of 5A-HBO and the solvent molecules.43,53 Even though it is clear that the basicity of the solvent plays a key role in the ESIPT reaction, we cannot completely rule out that the polarity of the solvent may affect the CT reaction and subsequently alter the ESIPT phenomenon.
Finally, the excitation spectra of DP-MPPI and DP-HPPI in the DCM and ACN solutions show similar bands to those obtained in the absorption ones (Fig. S3 and S4, ESI,† respectively), indicating that the emissive species originates from the same ground state. It is possible that these species (planar and nonplanar structures) coexist and get converted into the other in a very short time, both at S0 and S1 states. However, we do not have experimental evidence for this event.
Picosecond time-resolved fluorescence study
To unravel the ICT and ESIPT processes, we have examined the ps-photodynamics of DP-MPPI and DP-HPPI in the DCM and ACN solutions. Fig. 2 shows the emission decays of both molecules in DCM obtained after their photoexcitation using a 371 nm pulsed laser and the recorded signals at different wavelenghts (from 410 to 600 nm) of the emission spectra. Table 1 contains the time constants (τi), amplitudes (Ai) and preexponential factors (ai) normalized to 100, obtained from the multiexponential global analysis of emission decays.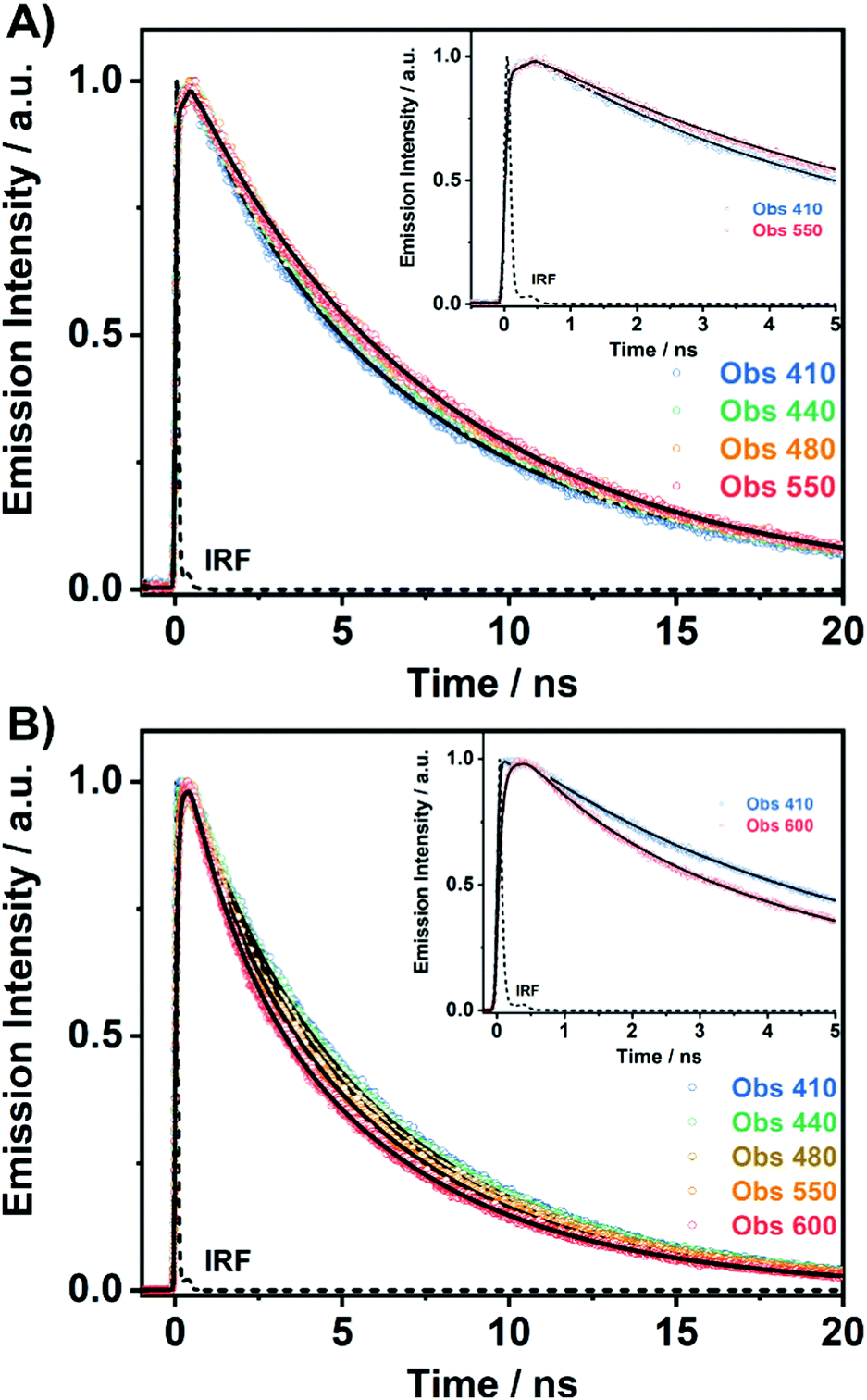 | ||
| Fig. 2 Magic-angle ps-emission decays of (A) DP-MPPI and (B) DP-HPPI in DCM solutions upon excitation at 371 nm. The observed wavelengths and a zoomed-in view of the decays are displayed in the inset. The solid lines are from the best fit using a multiexponential global function and IRF is the instrumental response function. | ||
| Sample | λ obs (nm) | τ 1/ps (±20) | a 1 | A 1 | τ 2/ns (±0.5) | a 2 | A 2 | τ 3/ns (±0.5) | a 3 | A 3 |
|---|---|---|---|---|---|---|---|---|---|---|
| DP-MPPI | 410 | — | — | — | 2.1 | 11 | — | 7.5 | 89 | — |
| 440 | — | — | 3 | — | 97 | — | ||||
| 480 | — | — | — | — | 100 | — | ||||
| 550 | — | — | — | — | 100 | — | ||||
| DP-HPPI | 410 | 90 | 18 | 335 | 1.5 | 1 | 94 | 6.0 | 81 | 1626 |
| 440 | 7 | 65 | 5 | 158 | 88 | 1483 | ||||
| 480 | −100 | −329 | 19 | 347 | 81 | 1236 | ||||
| 550 | −100 | −643 | 36 | 649 | 64 | 1050 | ||||
| 600 | −100 | −1033 | 40 | 991 | 60 | 862 |
For DP-MPPI, the emission decays were well fitted by biexponential analysis, giving two time constants: τ2 = 2.1 ns and τ3 = 7.5 ns (Fig. 2A and Table 1). The shortest τ2-component only contributes to the bluest part of the emission spectrum (410–440 nm) and disappears from wavelengths longer than 450 nm. On the other side, the longest τ3-component is present in the whole spectral region, increasing its contribution at the reddest part of the emission spectrum (Table 1). Considering these results and our previous discussion in the steady-state section, we suggest that this molecule undergoes an ultrafast ICT process (happening in shorter times than our ps-system resolution), leading to the formation of two different emitting CT species. Interestingly, the emission decay of MPPI exhibits a single exponential behaviour with a lifetime of 4.5 ns,47 and therefore it is clear that the phenyl groups in DP-MPPI create new pathways for its relaxation to the ground state. Attending to their chemical structure, the phenyl groups are able to rotate or twist around their axis, which may produce the coexistence of planar and non-planar conformers (Scheme 1). Generally, planar structures will emit light at longer wavelengths than non-planar ones because of their larger electronic conjugation. Based on this, and considering the contribution of each time component, it is reasonable to assign the shortest τ2-component to the emission lifetime of the non-planar CT conformers (emitting mostly at the bluest region of the spectrum), whereas the longest τ3-component is attributed to the emission lifetime of the planar CT species.
On the other hand, the photodynamics of DP-HPPI in DCM is more complex than that of DP-MPPI. The emission decays were well fitted to the multiexponential function, providing three time constants: τ1 = 90 ps, τ2 = 1.5 ns and τ3 = 6 ns (Fig. 2B and Table 1). The shortest component is decaying at the bluest region of the emission spectrum (410–440 nm), while it is rising at the reddest one (480–600 nm), reflecting the occurrence of a photoinduced process at S1. On the other hand, the intermediate (τ2) and longest (τ3) components decay in the whole spectral range. Note that the τ2-component (1.5 ns) increases its contribution with the observation wavelength, while that in DP-MPPI it disappears at wavelengths longer than 450 nm (Table 1), indicating that they have a different nature. In contrast, the longest τ3-component is comparable to the one obtained in DP-MPPI (7.5 ns), and, therefore, it can be attributed to the emission lifetime of the planar CT forms. Interestingly, the shortest (τ1) and intermediate (τ2) components show similar real amplitudes (Ai) but with opposite signs in the wavelengths corresponding to the emission of the K tautomer (600 nm, Table 1). This is a typical behaviour of a reversible two emitting state system, where the enol and K tautomers are in equilibrium (the classical Birks scheme, see in the ESI†). To shed more light on the reversibility of the ESIPT reaction in DP-HPPI, we recorded the time resolved emission spectra (TRES, Fig. 3). Some interesting conclusions can be extracted from this experiment: (i) the red-shifted emission band, attributed to the K emission, is formed within 80–100 ps, corroborating that the 90 ps (τ1) component is clearly associated with the ESIPT reaction; (ii) at longer delayed times, the emission intensities of the species emitting at ∼420 nm and keto (∼455 nm) tautomers are similar, indicating that both species are equilibrated at the excited state. It is worth noting that the parent HPPI molecule (without the 2 phenyl groups in the phenanthrene moiety) undergoes an ultrafast ESIPT reaction within 12 ps.47 Hence, the introduction of the electron-rich phenyl groups in DP-HPPI, which induces the ICT reaction, is at the same time slowing down the ESIPT phenomenon. It is well-known that the ICT reaction will produce a charge reorganization thereby affecting the acidity/basicity of the proton donor and acceptor moieties of the molecules, which may cause a slowing down of the ESIPT process. For instance, the amino-functionalized 6A-HBO and 5A-HBO molecules have shown an abnormally slow proton transfer (up to 15–20 ps) compared to the parent molecule HBO (less than 150 fs).43,50,51,53 Therefore, the ultrafast ICT reaction induced charge reorganization affecting the driving force of the proton donor and acceptor groups, reducing in this way the velocity of the ESIPT reaction.
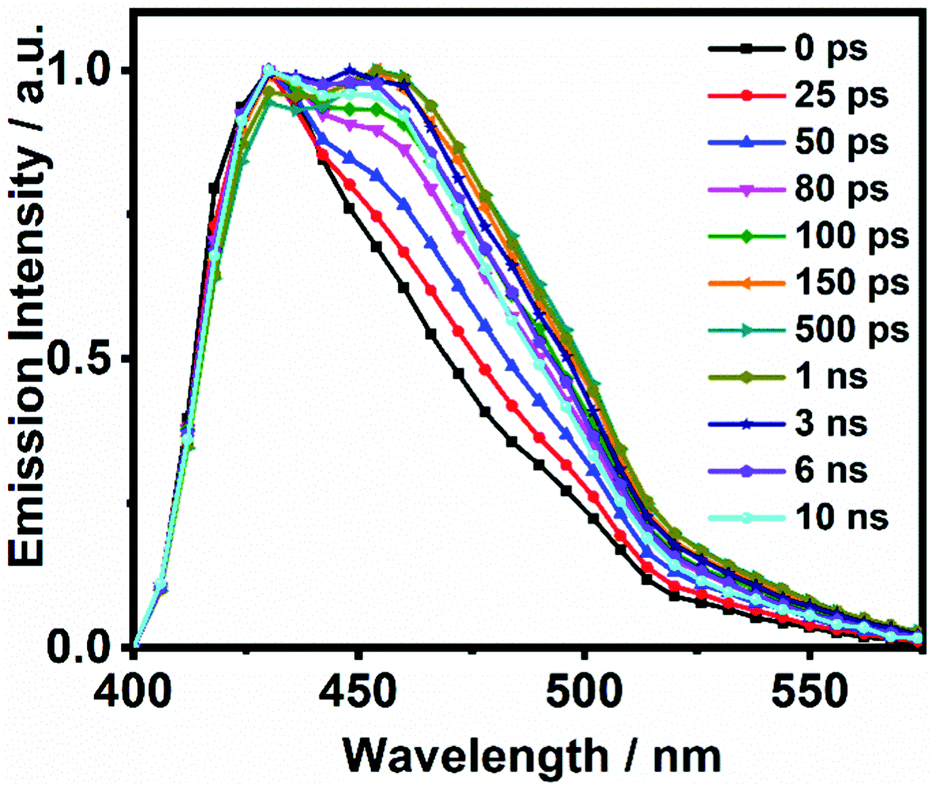 | ||
| Fig. 3 Normalized time-resolved emission-spectra (TRES) of DP-HPPI in DCM upon excitation at 371 nm and gating at the indicated delay times. | ||
To obtain the values of the forward and reverse proton-transfer processes linking these structures and the fluorescence lifetime of the involved species, we analysed the emission decays according to the classical Birks for a 2-state model involving two structures in equilibrium at S1, using the equations reported elsewhere,54–56 and shown in the ESI.† Following the analysis, we obtained the value of the emission lifetime of the K tautomer (τK = 1.4 ns), the rate constants of the direct (kDPT = 8.35 × 109 s−1) and reverse proton transfer (kRPT = 2.24 × 109 s−1) reactions, and the time of both events: τDPT = 120 ps and τRPT = 447 ps. Note that the direct proton transfer (DPT) reaction occurs within a shorter time than that of the reverse one (RPT), indicating that the former reaction is favoured over the second one. The equilibrium constant K (kDPT/kRPT) is 3.72, while ΔG0(293 K) = −3.19 kJ mol−1. Comparable results were reported for some HBO derivatives functionalized with an electron donating amino group.43,57,58 For instance, 6-amino-2-(2-hydroxyphenyl)benzoxazole (6A-HBO) in DCM undergoes the ultrafast ICT reaction (∼80 fs) followed by the reversible ESIPT process, being the time constants for the direct and reverse reactions, τDPT = 129 ps and τRPT = 335 ps, the equilibrium constant K = 2.57, and ΔG0(293 K) = −2.3 kJ mol−1.57 While the time constant of DPT is very similar (120–130 ps), the time constant of RPT differs by 110 ps, being shorter in 6A-HBO. These results suggest that the activation energies for DPT are very similar for both molecules, while they differ in 0.73 kJ mol−1 for RPT, being the lowest one for 6A-HBO.
We have also investigated the photobehavior of these molecules in ACN. The emission decays are displayed in Fig. S5 (ESI†), while the values of the time constants (τi) and pre-exponential factors (ai) normalized to 100 obtained from the single or multiexponential global fit analysis are shown in Table S1 (ESI†). The emission decays of DP-MPPI were well-fitted to the monoexponential function, giving a time constant of 7 ns. According to our previous assignments, this component can be attributed to the emission lifetime of the planar CT species.
On the other hand, for DP-HPPI, biexponential analysis was required for an accurate fit of the decay traces, providing two time constants: τ1 = 2 ns and τ2 = 6.9 ns. Both components are decaying in the whole spectral range. The longest τ2-component shows its maximum contribution at 410 nm (95%) which then decreases at longer observation wavelengths, while the shortest τ1-one shows a relatively weak contribution at 410 nm (5%), increasing towards longer wavelengths. The lack of a rise signal on DP-HPPI in the ACN solution corroborates the absence of the ESIPT reaction due to the intermolecular interactions between the proton of the molecule and the solvent, which is in agreement with our previous discussion. Hence, the only emitting species of DP-HPPI in this solvent are the CT ones. The longest τ2-component (6.9 ns) is assigned to the planar CT conformer, while the shortest τ2-one (2 ns) corresponds to the emission lifetime of the non-planar CT form. The absence of the ESIPT reaction is also endorsed by the TRES, in which there is a lack of the red-shifted emission band corresponding to the emission of the K species (Fig. S6, ESI†).
Femtosecond dynamics
To obtain precise information on the time constant of the ICT reaction, we have performed fs-experiments on the ultrafast excited state photodynamics of both molecules in DCM. We used the fs-up conversion technique. The samples were pumped using a 320 nm fs-laser and probed at different emission wavelengths. The obtained transients are displayed in Fig. 4, while the obtained time constants (τi) and pre-exponential factors (ai) normalized to 100 are given in Table 2.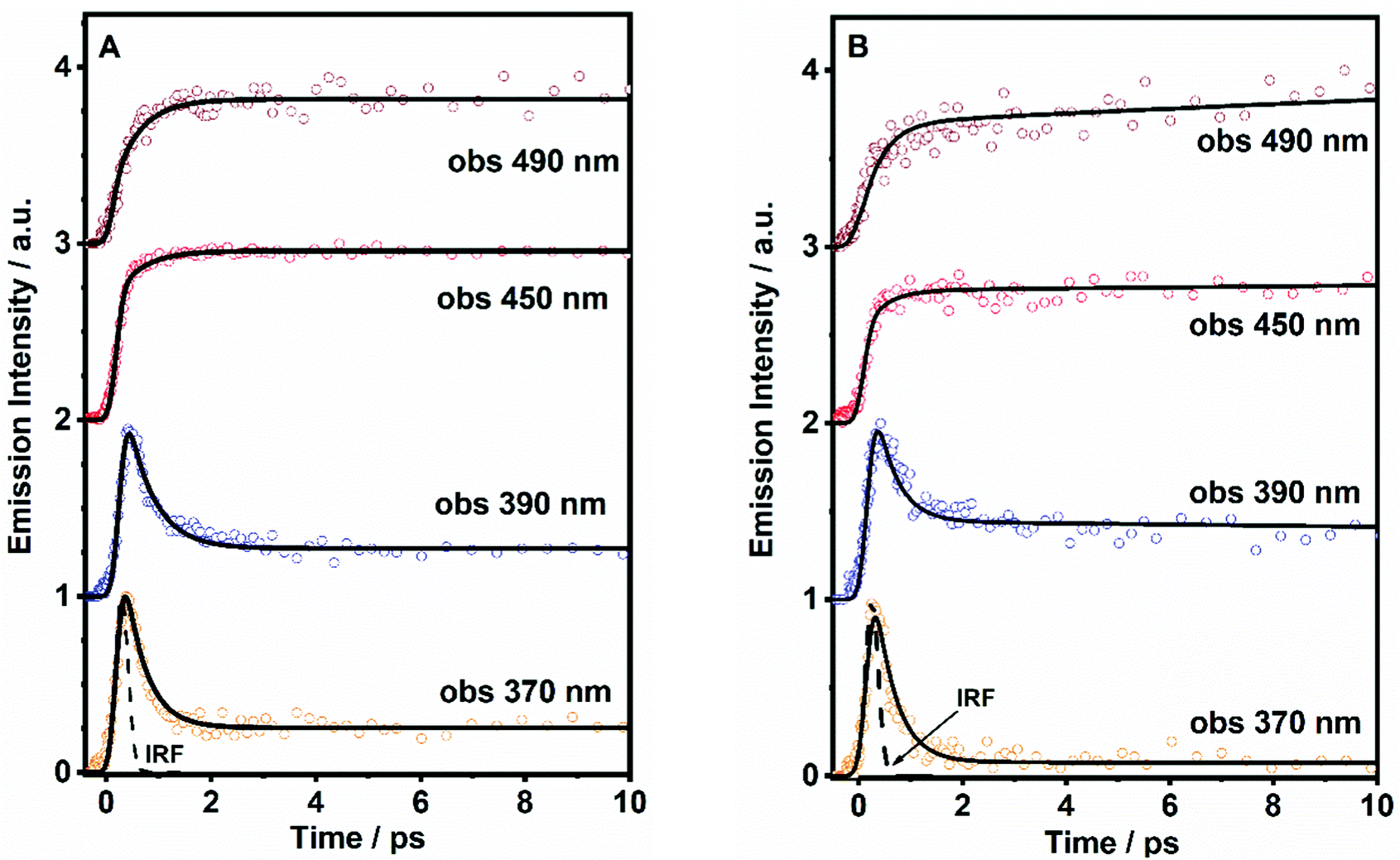 | ||
| Fig. 4 Representative fs-emission transients of (A) DP-MPPI and (B) DP-HPPI in DCM solutions upon excitation at 320 nm and probing at the indicated wavelengths. The solid lines are from the best fit using a multiexponential function. IRF is the instrumental response function. | ||
| Sample | λ Obs (nm) | τ 1/fs (±100) | a 1 | τ 2/ps (±10) | a 2 | τ 3/ns (±0.5) | a 3 |
|---|---|---|---|---|---|---|---|
| DP-MPPI | 370 | 360 | 84 | — | — | 7* | 16 |
| 390 | 520 | 78 | — | — | 22 | ||
| 450 | 400 | −100 | — | — | 100 | ||
| 490 | 550 | −100 | — | — | 100 | ||
| DP-HPPI | 370 | 350 | 97 | 67 | 2 | 5.8* | 1 |
| 390 | 700 | 70 | 68 | 15 | 15 | ||
| 450 | 750 | −48 | 70 | −52 | 100 | ||
| 600 | 800 | −29 | 70 | −71 | 100 |
The analysis of the decays of DP-MPPI provides two time components, an ultrafast component of ∼400 fs and an offset component of 7 ns (fixed from the values obtained from the TCSPC experiments, Fig. 4A and Table 2). The ultrafast 400 fs-component decays at the very bluest part of the emission spectrum (370–390 nm), while it rises from wavelengths longer than 450 nm. This clearly evidences the existence of the ultrafast reaction in the excited state of this molecule, and considering our previous observations, this reaction can be attributed to the ICT process, as this molecule cannot undergo the ESIPT event. These kinds of ultrafast ICT reactions have also been described for other benzoxazoles derivatives, supporting our argument.50,57 For example, the 6-amino-2-(2′-methoxyphenyl)benzoxazole (6A-MBO) molecule has undergone the ultrafast ICT process happening in 150 fs.50,57
On the other hand, the ultrafast emission decay of DP-HPPI exhibits the multiexponential behaviour with time constants of τ1 = 700 fs and τ2 = 70 ps, and an offset of 5.8 ns (fixed from the values obtained from TCSPC) as shown in Fig. 4B, Fig. S7 (ESI†), and Table 2. The τ2-component is comparable to the 90 ps detected in the ps-TCSPC experiments, decaying at the bluest part, and increasing at the reddest region. Hence, following our previous discussion, this component is associated with the direct/reverse proton-transfer reactions. The ultrafast τ1-component (700 fs) behaves very similar to that observed for DP-MPPI, decaying at the very bluest emission region, and increasing from 450 nm and above. In this sense, this ultrafast component can be assigned to the excited-state ICT reaction, which is in agreement with all our previous observations.
Considering all the results obtained up to now, we can offer a global and precise vision of the photodynamics scheme of DP-MPPI and DP-HPPI molecules in DCM and ACN solvents. As illustrated in Scheme 2A, the photoexcited DP-MPPI in the DCM molecule undergoes an ultrafast ICT reaction (400 fs), with the subsequent formation of two different CT species, with non-planar and planar conformations that emit with lifetimes of 2.1 and 7.5 ns, respectively. On the other hand, the photodynamics of DP-HPPI strongly depends on the properties of the solvent. In DCM, it undergoes an ultrafast ICT reaction (700 fs), followed by a reversible ESIPT reaction with the emission of K forms, non-planar ECT and planar ECT species in 1.4, 2.1 and 6 ns, respectively (Scheme 2B). However, in ACN (the more basic and polar solvent), this molecule does not show any signature of the ESIPT reaction due to intermolecular interactions with the solvent molecules, so its photophysical behaviour resembles that of DP-MPPI, with an ultrafast ICT reaction and the subsequent formation of non-planar and planar CT species emitting with lifetimes of 2.1 and 6 ns.
 | ||
| Scheme 2 Proposed photodynamic schemes for (A) DP-MPPI and (B) DP-HPPI in DCM solutions, in which ICT is the intramolecular charge transfer process and τDPT and τRPT are the times for the direct and reverse ESIPT reactions, respectively. | ||
Conclusions
In this work, we have investigated the effect of phenyl groups in the proton accepting moiety of a new proton transfer molecule (DP-HPPI) and its methylated equivalent (DP-MPPI) on their photophysical properties. First, the large Stokes shift observed in the steady state emission spectra reflects the existence of ICT, which is induced by the phenyl groups in the phenanthrene moiety. Second, the dual emission of DP-HPPI in DCM indicates the subsequent occurrence of an ESIPT reaction which was precluded in ACN owing to the more polar and basic properties of this solvent, which favours interactions with the proton atom of DP-HPPI. The fs–ps experiments on both molecules support the assignments and provide the time constants of ultrafast events. We observed the existence of an ultrafast process in the excited state (400–700 fs) which is attributed to the ICT reaction, leading to the formation of non-planar and planar CT species with lifetimes of 2 and 6–7.5 ns, respectively. We have also proved that DP-HPPI in the DCM solution undergoes a subsequent fast reversible ESIPT reaction, leading to the formation of a K tautomer which is in equilibrium with the enol CT species. These results highlight the influence of phenyl groups in the tunability of the emission and photophysical properties of proton transfer dyes, opening a way for a controlled design of new molecules and systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants: PID2020-116519RB-I00, MAT2017-82288-C2-2-P, and PID2020-112590GB-C22 funded by MCIN/AEI/10.13039/501100011033 and by the European Union (EU); SBPLY/19/180501/000212 funded by JCCM and by the EU through “Fondo Europeo de Desarollo Regional” (FEDER); and 2020-GRIN-28929 funded by UCLM (FEDER). M. G. also thanks the EU for financial support through Fondo Social Europeo Plus (FSE+).Notes and references
- E. M. Kosower and D. Huppert, Annu. Rev. Phys. Chem., 1986, 37(1), 127–156 CrossRef CAS.
- A. Douhal, S. K. Kim and A. H. Zewail, Nature, 1995, 378(6554), 260–263 CrossRef CAS PubMed.
- A. Douhal, F. Lahmani and A. Zehnacker-Rentien, Chem. Phys., 1993, 178(1-3), 493–504 CrossRef CAS.
- J. T. Hynes, T.-H. Tran-Thi and G. Granucci, J. Photochem. Photobiol., A, 2002, 154(1), 3–11 CrossRef CAS.
- V. I. Tomin, Proton Transfer Reactions in the Excited Electronic State, Hydrogen Bonding and Transfer in the Excited State, John Wiley & Sons, Ltd, 2010, pp 463–523 Search PubMed.
- D. P. Zhong, A. Douhal and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(26), 14056–14061 CrossRef CAS PubMed.
- M. Kasha, J. Chem. Soc., Faraday Trans. 2, 1986, 82(12), 2379–2392 RSC.
- V. I. Tomin, A. P. Demchenko and P.-T. Chou, J. Photochem. Photobiol., C, 2015, 22, 1–18 CrossRef CAS.
- H. Wang, H. Zhang, O. K. Abou-Zied, C. Yu, F. E. Romesberg and M. Glasbeek, Chem. Phys. Lett., 2003, 367(5–6), 599–608 CrossRef CAS.
- I. Belevich, M. I. Verkhovsky and M. Wikström, Nature, 2006, 440(7085), 829–832 CrossRef CAS PubMed.
- A. R. Crofts, Biochim. Biophys. Acta, 2004, 1655, 77–92 CrossRef CAS PubMed.
- S. J. Formosinho and L. G. Arnaut, J. Photochem. Photobiol., A, 1993, 75(1), 21–48 CrossRef CAS.
- A. Douhal, F. Lahmani and A. H. Zewail, Chem. Phys., 1996, 207(2–3), 477–498 CrossRef CAS.
- A. U. Acuna, A. Costela and J. M. Munoz, J. Phys. Chem., 1986, 90(13), 2807–2808 CrossRef CAS.
- C.-C. Yan, X.-D. Wang and L.-S. Liao, ACS Photonics, 2020, 7(6), 1355–1366 CrossRef CAS.
- H. Yin, Y.-M. Zhang, H.-F. Zhao, G. Yang, Y. Shi, S. X.-A. Zhang and D.-J. Ding, Dyes Pigm., 2018, 159, 506–512 CrossRef CAS.
- Y. Zhang, H. Yang, H. Ma, G. Bian, Q. Zang, J. Sun, C. Zhang, Z. An and W.-Y. Wong, Angew. Chem., Int. Ed., 2019, 58(26), 8773–8778 CrossRef CAS PubMed.
- Q. Zhang, L. Yang, Y. Han, Z. Wang, H. Li, S. Sun and Y. Xu, Chem. Eng. J., 2022, 428, 130986 CrossRef CAS.
- P. Zhou and K. Han, Acc. Chem. Res., 2018, 51(7), 1681–1690 CrossRef CAS PubMed.
- M. Shahid and A. Misra, J. Photochem. Photobiol., A, 2017, 335, 190–199 CrossRef CAS.
- M. Mohapatra and A. K. Mishra, Photochem. Photobiol. Sci., 2019, 18(12), 2830–2848 CrossRef CAS PubMed.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47(23), 8842–8880 RSC.
- S. Park, S. Kim, J. Seo and S. Y. Park, Macromol. Res., 2008, 16(5), 385–395 CrossRef CAS.
- W. Sun, S. Li, R. Hu, Y. Qian, S. Wang and G. Yang, J. Phys. Chem. A, 2009, 113(20), 5888–5895 CrossRef CAS PubMed.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23(32), 3615–3642 CrossRef CAS PubMed.
- H. C. Joshi and L. Antonov, Molecules, 2021, 26(5), 1475 CrossRef CAS PubMed.
- C. Jouvet, M. Miyazaki and M. Fujii, Chem. Sci., 2021, 12(11), 3836–3856 RSC.
- C.-H. Wu, L. J. Karas, H. Ottosson and J. I. C. Wu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(41), 20303 CrossRef CAS PubMed.
- Q. Huang, Q. Guo, J. Lan and J. You, Dyes Pigm., 2021, 193, 109497 CrossRef CAS.
- Y. Shen, X. Liu, X. Zhang, Y. Zhang and B. Gu, Spectrochim. Acta, Part A, 2020, 239, 118515 CrossRef CAS PubMed.
- M. Tao, L. Wen, D. Huo, Z. Kuang, D. Song, Y. Wan, H. Zhao, J. Yan and A. Xia, J. Phys. Chem. B, 2021, 125(40), 11275–11284 CrossRef CAS PubMed.
- K. Das, N. Sarkar, A. K. Ghosh, D. Majumdar, D. N. Nath and K. Bhattacharyya, J. Phys. Chem., 1994, 98(37), 9126–9132 CrossRef CAS.
- K. Das, N. Sarkar, D. Majumdar and K. Bhattacharyya, Chem. Phys. Lett., 1992, 198(5), 443–448 CrossRef CAS.
- S. Lochbrunner, A. J. Wurzer and E. Riedle, J. Phys. Chem. A, 2003, 107(49), 10580–10590 CrossRef CAS.
- C. A. S. Potter and R. G. Brown, Chem. Phys. Lett., 1988, 153(1), 7–12 CrossRef CAS.
- O. K. Abou-Zied, Chem. Phys., 2007, 337(1–3), 1–10 CrossRef CAS.
- O. K. Abou-Zied, R. Jimenez, E. H. Z. Thompson, D. P. Millar and F. E. Romesberg, J. Phys. Chem. A, 2002, 106(15), 3665–3672 CrossRef CAS.
- A. Douhal, F. Amat-Guerri, M. P. Lillo and A. U. Acuna, J. Photochem. Photobiol., A, 1994, 78(2), 127–138 CrossRef CAS.
- M. Sanz and A. Douhal, Chem. Phys. Lett., 2005, 401(4–6), 435–439 CrossRef CAS.
- F. S. Santos, E. Ramasamy, V. Ramamurthy and F. S. Rodembusch, J. Photochem. Photobiol., A, 2016, 317, 175–185 CrossRef CAS.
- S. M. Aly, A. Usman, M. AlZayer, G. A. Hamdi, E. Alarousu and O. F. Mohammed, J. Phys. Chem. B, 2015, 119(6), 2596–2603 CrossRef CAS PubMed.
- G. J. Woolfe, M. Melzig, S. Schneider and F. Dörr, Chem. Phys., 1983, 77(2), 213–221 CrossRef CAS.
- N. Alarcos, M. Gutierrez, M. Liras, F. Sanchez, M. Moreno and A. Douhal, Phys. Chem. Chem. Phys., 2015, 17(22), 14569–14581 RSC.
- C. H. Kim, J. Park, J. Seo, S. Y. Park and T. Joo, J. Phys. Chem. A, 2010, 114(18), 5618–5629 CrossRef CAS PubMed.
- S. Ríos Vázquez, M. C. Ríos Rodríguez, M. Mosquera and F. Rodríguez-Prieto, J. Phys. Chem. A, 2007, 111(10), 1814–1826 CrossRef PubMed.
- S. Ríos Vázquez, M. C. Ríos Rodríguez, M. Mosquera and F. Rodríguez-Prieto, J. Phys. Chem. A, 2008, 112(3), 376–387 CrossRef PubMed.
- E. Gomez, N. Alarcos, C. Monterde, F. Sánchez, M. Moreno and A. Douhal, Phys. Chem. Chem. Phys., 2018, 20(42), 27149–27161 RSC.
- J. A. Organero, L. Tormo and A. Douhal, Chem. Phys. Lett., 2002, 363(3), 409–414 CrossRef CAS.
- M. Gil and A. Douhal, Chem. Phys. Lett., 2006, 428(1–3), 174–177 CrossRef CAS.
- N. Alarcos, M. Gutierrez, M. Liras, F. Sanchez and A. Douhal, Phys. Chem. Chem. Phys., 2015, 17(25), 16257–16269 RSC.
- M. Gutierrez, N. Alarcos, M. Liras, F. Sánchez and A. Douhal, J. Phys. Chem. B, 2015, 119(2), 552–562 CrossRef CAS PubMed.
- Y.-M. Cheng, S.-C. Pu, C.-J. Hsu, C.-H. Lai and P.-T. Chou, Chem. Phys. Chem., 2006, 7(6), 1372–1381 CrossRef CAS PubMed.
- N. Alarcos, M. Gutierrez, M. Liras, F. Sanchez and A. Douhal, Photochem. Photobiol. Sci., 2015, 14(7), 1306–1318 CrossRef CAS PubMed.
- S. I. Druzhinin, N. P. Ernsting, S. A. Kovalenko, L. P. Lustres, T. A. Senyushkina and K. A. Zachariasse, J. Phys. Chem. A, 2006, 110(9), 2955–2969 CrossRef CAS PubMed.
- Y. V. Il'ichev, W. Kühnle and K. A. Zachariasse, J. Phys. Chem. A, 1998, 102(28), 5670–5680 CrossRef.
- K. A. Zachariasse, T. Yoshihara and S. I. Druzhinin, J. Phys. Chem. A, 2002, 106(26), 6325–6333 CrossRef CAS.
- M. Gutierrez, N. Alarcos, M. Liras, F. Sánchez and A. Douhal, J. Phys. Chem. B, 2015, 119(2), 552–562 CrossRef CAS PubMed.
- A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42(3), 1379–1408 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Scheme S1 shows the steps followed in the synthesis of the DP-MPPI and DP-HPPI molecules. Fig. S1 and S2 display the 1H NMR (in DMSO-d6) and FTIR spectra (in KBr) of DP-MPPI and DP-HPPI, respectively. Fig. S3 and S4 present the excitation spectra of DP-MPPI and DP-HPPI in DCM and ACN solutions, respectively. Fig. S5 shows the magic-angle emission decay of DP-MPPI and DP-HPPI excited at 371 nm in ACN, while Fig. S6 displays the TRES of DP-HPPI in ACN solution. Fig. S7 shows the fs-emission transients of DP-HPPI in DCM solution upon excitation at 320 nm with a long temporal window. Finally, Table S1 shows the values of time constants (τi) and normalized (to 100) pre-exponential factors (ai) obtained from a multiexponential global fit analysis of the emission decays of DP-MPP and DP-HPP in ACN solutions upon excitation at 371 nm. See DOI: 10.1039/d1cp05294b |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2022 |