
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthetic routes to bridge-functionalised calix[4]arenes†
Angela
Fong
a,
Cameron L.
Campbell
 a,
Silvia
Huynh
a,
Laura J.
McCormick McPherson
b,
Simon J.
Teat
b,
Magnus W. P.
Bebbington
ac and
Scott J.
Dalgarno
*a
a,
Silvia
Huynh
a,
Laura J.
McCormick McPherson
b,
Simon J.
Teat
b,
Magnus W. P.
Bebbington
ac and
Scott J.
Dalgarno
*a
aInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh, EH14 4AS, UK. E-mail: S.J.Dalgarno@hw.ac.uk
bStation 11.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, USA
cDepartment of Chemistry and Biochemistry, Montclair State University, 1 Normal Ave., Montclair, NJ 07043, USA
First published on 3rd February 2022
Abstract
Ring-opening of furans at the equatorial methylene bridge positions of a calix[4]arene gives access to a range of new molecules (in good yield) that have widespread potential impact in supramolecular chemistry amongst other areas.
Calix[n]arenes are cyclic host molecules (general notation C[n]s herein, where n represents the number of aryl rings present) that have played a pivotal role in the development of supramolecular chemistry over the past 30–40 years. Reasons for such widespread application include (but are not limited to) tuneable ring size,1 structural flexibility associated with conformational interconversion,2 cavities or clefts that can house suitable guest species,3 and versatility towards synthetic alteration at the upper- or lower-rims.4–7 Of the family of C[n]s, the cyclic tetramers have arguably been most widely studied as their conformation can be readily controlled.8 In addition, synthetic modification of C[4]s at either the upper- or lower-rims (or both) is well explored.9,10 Although this is the case, the synthesis of exhaustively methylene bridge-functionalised species remains a significant challenge.11,12 Access to a wide range of such species would be of tremendous benefit as one could exploit many of the characteristic C[4] properties whilst simultaneously imparting new functionality on the periphery of this extremely versatile scaffold. This contribution addresses this challenge in part, presenting a new and versatile route to exhaustively methylene bridge-functionalised C[4]s.
Access to C[4] chemistry is typically via a one-pot synthesis of p-tert-butylcalix[4]arene (TBC[4]) from p-tert-butylphenol and formaldehyde under well-established reaction conditions.13 Literature concerned with synthetic C[4] upper-/lower-rim alteration is vast, but reports of functionalisation at all methylene bridge positions are limited. Several examples have shown substitution at one or two positions around the C[4] framework11,12,14,15 and in some cases this methodology has been used to tether C[4]s with linkers of varying length and/or composition (e.g. alkyl vs. aryl linkers).16 However, mono-substitution at all methylene bridges presents a major challenge as these groups can be introduced either axial or equatorial (Fig. 1); this can give rise to isomers that can be difficult to separate. Rather than explore this particular step further, we sought to utilise a previously reported compound in which all four groups at the methylene bridge were known to occupy equatorial positions, thus allowing us to preserve the C[4] cone conformation whilst undertaking subsequent synthetic transformations. The compound in question, 5,11,17,23-tetra-tert-butyl-2,8,14,20-tetrakis(2-methylfuranyl)-25,26,27,28-tetramethoxycalix[4]arene (1, Fig. 1), was previously reported by Biali and co-workers and is isolated by methylation of the lower-rim of TBC[4],17 mono-bromination at each methylene bridge, and solvolytic reaction with 2-methylfuran in the presence of butylene oxide. The authors reported a Diels–Alder reaction of 1 with benzyne to afford a tetra-naphthyl derivative, but to our knowledge this exhaustively bridge-substituted calixarene has not been further exploited.
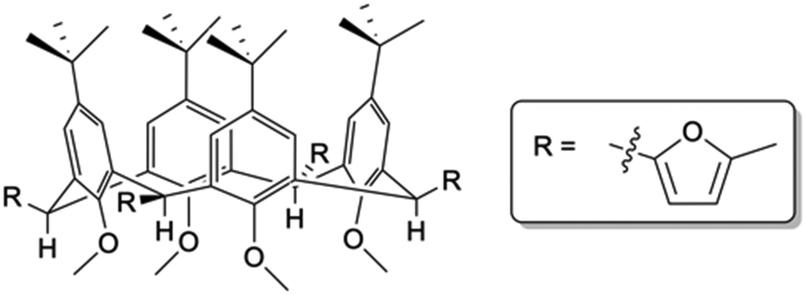 | ||
| Fig. 1 Schematic of 1 showing 2-methylfuranyl moieties monosubstituted equatorially at all four methylene bridge positions of the TBC[4] framework. | ||
Ring-opening of the furan moieties in 1 to afford either unsaturated or saturated 1,4-diketones gives numerous possibilities for the introduction of functionalities to the C[4] methylene bridge. Here we report formation of both diketones, but also their use in the synthesis of a range of C[4]s that are mono-substituted at all four methylene bridges. We also report preliminary work in lower-rim deprotection following alteration of methylene bridge functionality, as reintroducing hydroxyl groups in that region of the scaffold will be immensely beneficial (e.g. for use in coordination chemistry whilst also exploiting peripheral groups present at the methylene bridges).
Introduction of a 2-methylfuranyl moiety at each C[4] methylene bridge position presents a range of opportunities for synthetic transformation. Furans are known as synthetically diverse species that can be modified through various reactions including electrophilic aromatic substitution, Diels–Alder and nucleophilic addition.18,19 One reaction of interest to us was ring-opening,19,20 as it provides the opportunity to isolate other equatorial heterocycles at each C[4] methylene bridge. This was achieved by oxidation using a peroxy acid (mCPBA) or via acid-catalysed hydrolysis, affording unsaturated or saturated diketones respectively (2 and 3, Scheme 1).
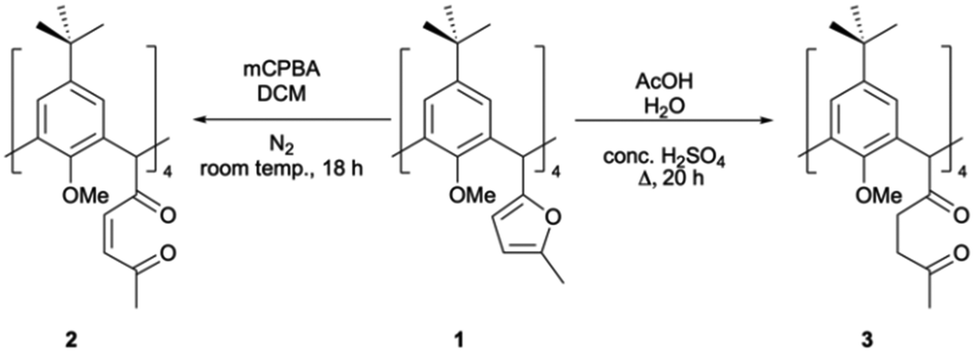 | ||
| Scheme 1 Synthesis of C[4]s with unsaturated (2) and saturated (3) diketones at each methylene bridge of a p-tert-butylcalix[4]arene. | ||
Reaction of 1 with mCPBA in DCM afforded 2 in 34% yield upon purification by column chromatography (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 EtOAc/PET). Colourless single crystals were grown by recrystallisation from acetonitrile and the structure of 2 is shown in Fig. 2A. Each furan moiety on the C[4] scaffold has been ring-opened as indicated by four carbon–carbon bond lengths (in the 1,4-diketone backbones) in the range of 1.318(5)–1.325(5) Å. The presence of carbonyl groups was also confirmed by both IR and 13C NMR spectroscopies, with introduction of a band at 1696 cm−1 and signals at 201.5 and 199.9 ppm respectively.
:2 EtOAc/PET). Colourless single crystals were grown by recrystallisation from acetonitrile and the structure of 2 is shown in Fig. 2A. Each furan moiety on the C[4] scaffold has been ring-opened as indicated by four carbon–carbon bond lengths (in the 1,4-diketone backbones) in the range of 1.318(5)–1.325(5) Å. The presence of carbonyl groups was also confirmed by both IR and 13C NMR spectroscopies, with introduction of a band at 1696 cm−1 and signals at 201.5 and 199.9 ppm respectively.
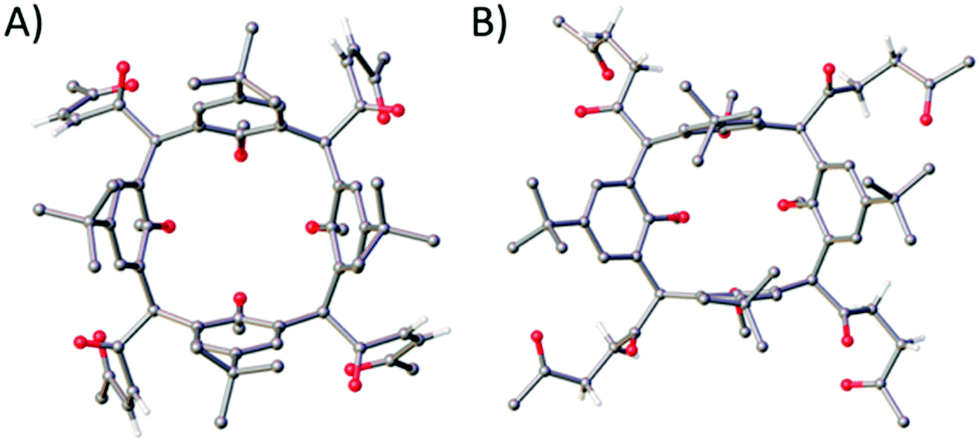 | ||
| Fig. 2 Top down views of single crystal X-ray structures of C[4]s with unsaturated (2) and saturated (3) diketones at the methylene bridge positions. Colour code: C – grey, H – white, O – red. H atoms (except those in the diketone backbones) and solvent of crystallisation omitted for clarity. Figures not to scale. | ||
Alternatively, heating 1 at reflux in a mixture of acetic acid, conc. sulfuric acid and water for 20 h, followed by purification via column chromatography (7:3 CHCl3/EtOAc) afforded 3 in 53% yield. Colourless single crystals of 3 were grown from methanol, the structure of which is shown in Fig. 2B. Inspection of the structure shows that the furan group has been ring-opened to give the saturated diketone species, with C–C bond lengths (of the diketone backbone) in the range of 1.508(2)–1.5191(18) Å. Compound 3 has an extremely useful NMR handle associated with the methylene groups of the saturated diketones. These present as two very distinct triplets in the 1H NMR at 2.81 and 2.91 ppm and can be used to monitor consumption of 3 in further reactions (vide infra). As for 2, the presence of carbonyl groups in 3 was also confirmed by both IR and 13C NMR spectroscopies, with the introduction of a band at 1713 cm−1 and signals at 208.8 and 207.4 ppm respectively.
With these new compounds in hand, we began to explore exhaustive formation of new heterocycles at the methylene bridge positions. Starting with 2, reaction with hydrazine hydrate in acetic acid at reflux gave the tetra pyridazine analogue 4 in 67% yield. IR spectroscopy conveniently confirmed reaction completion upon consumption of the unsaturated diketones at the methylene bridges. Single crystals of 4 were grown by vapour diffusion of chloroform/acetonitrile and the structure is shown in Fig. 3. Inspection of the structure shows successful formation of a pyridazine moiety at each bridge and retention of equatorial positioning. Following the successful isolation of 4, we attempted to react 2 to form the tetrathiophene C[4] analogue,21 however all attempts were unsuccessful and returned starting material.
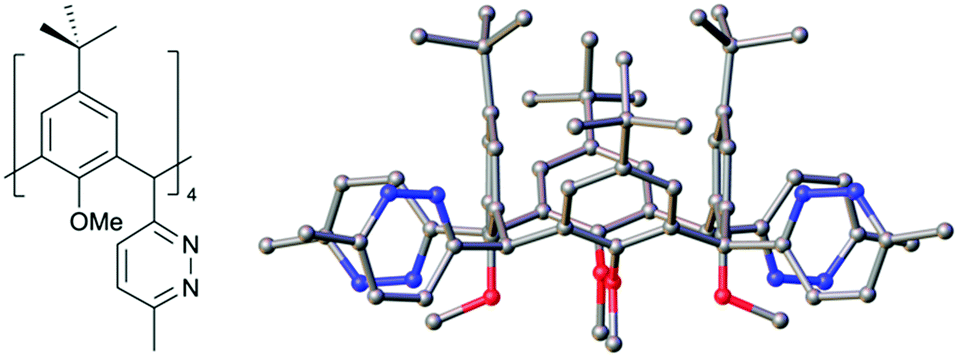 | ||
| Fig. 3 Schematic and side-on view of the single crystal X-ray structure of 4. Colour code: C – grey, N – blue, O – red. | ||
We expected 3 to be a more versatile reagent, as the saturated diketones can be exploited in the formation of a vast array of N-substituted pyrrole analogues through Paal–Knorr synthesis.22 We selected the reagents in Fig. 4A as a starting point with a view to forming pyrroles 5A–E (Fig. 4B).
 | ||
| Fig. 4 Reagents (A) selected for reaction with 3 with the aim of isolating tetra pyrrolic C[4]s 5A–E (B). | ||
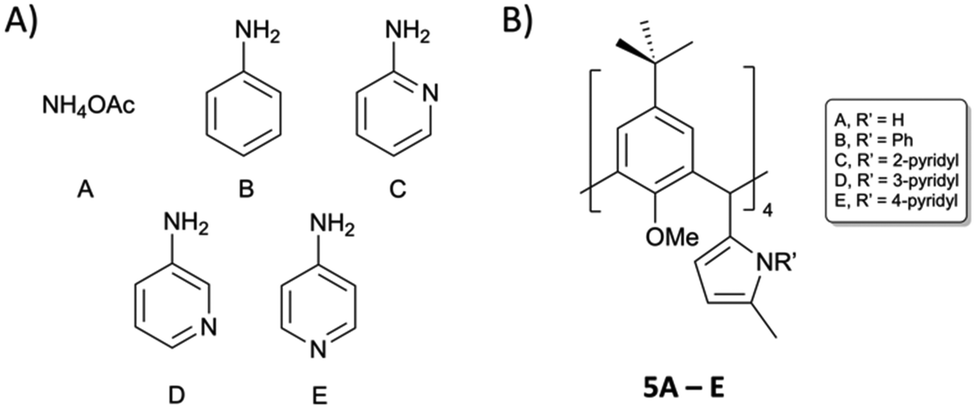
Reaction of 3 with ammonium acetate in acetic acid afforded compound 5A in 69% yield following purification by column chromatography (9:1 CHCl3/EtOAc), which is appreciable when one considers the reaction is occurring four times per C[4] scaffold. Inspection of the 1H NMR spectrum showed loss of the 1,4-diketone NMR handle with concomitant introduction of a set of doublets at 5.82 ppm and a broad singlet at 7.68 ppm corresponding to the CH and NH protons of the pyrrole moiety, respectively. Colourless single crystals of 5A were grown from vapour diffusion of methanol into a saturated DCM solution and structural analysis also confirmed successful Paal–Knorr pyrrole synthesis (Fig. 5A). Inspection of the structure of 5A showed that all pyrrole groups remained equatorial as expected, and that the C[4] is in a pinched cone conformation.
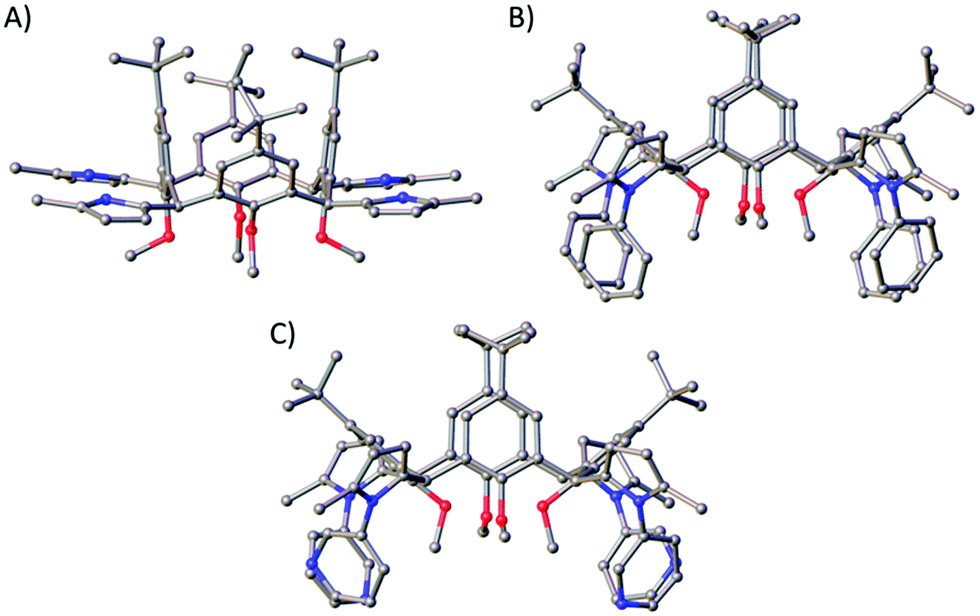 | ||
| Fig. 5 Single crystal X-ray structures of 5A (A), 5B (B) and 5D (C). Colour code: C – grey, N – blue, O – red. H atoms and solvent of crystallisation omitted for clarity. Figures not to scale. | ||
Reaction of 3 with aniline at reflux in toluene and in the presence of p-toluenesulfonic acid monohydrate (p-TsOH·H2O) for 24 hours resulted in formation of 5B in 55% yield. As in the synthesis of 5A, loss of the signals for the 1,4-diketone NMR handle occurred with the introduction of a set of doublets at 5.93 and 5.99 ppm corresponding to the CH protons of the pyrrole moieties in 5B, and multiple additional peaks in the aromatic region for the N-phenyl groups. The latter signals were broad, a feature that may be related to hindered rotation or C[4] conformational locking due to the bulk of the N-phenyl groups at the methylene bridge. Unfortunately, it was not possible to study this further (e.g. through VT experiments) as 5B has limited solubility. It was, however, possible to obtain colourless single crystals of 5B upon crystallisation from CHCl3, in which it is sparingly soluble. Subsequent structural analysis confirmed pyrrole formation at each methylene bridge, with the C[4] again adopting a pinched-cone conformation (Fig. 5B).
Following successful formation of 5A/B, we turned our attention to aminopyridines as these would afford interesting scaffolds for coordination chemistry/metal-directed assembly (e.g. to afford metal–organic cages23). Replacement of aniline with 2-, 3-, or 4-aminopyridine (2-, 3-, or 4-AP respectively) in the above reaction was successful in only one case. Reaction of 3 with 3-AP at reflux for seven days afforded 5D a white solid in 55% yield following purification by column chromatography (9:1 DCM/MeOH). As was the case for 5B, the RT 1H NMR spectrum of 5D also displayed broad aromatic signals. This was again thought to be due to hindered rotation of the pyridine moieties, but in this case it was possible to carry out VT NMR analysis to investigate whether the compound would display thermally induced rotation of the pyridine moieties (and therefore lead to much sharper and distinct NMR signals). Upon warming from 25 °C to 75 °C it was possible to see significant peak sharpening, and it is interesting to note the broad peaks around 8.4 and 8.0 ppm coalesce as the temperature is raised. This peak would be expected to be much sharper and distinct if heating beyond 75 °C, but it was not possible to reach higher temperatures. The same phenomenon occurred for the two broad peaks at 7.58 and 6.99 ppm, and again, much sharper signals would be expected if higher temperatures were accessible. This process is reversible, and signal broadening is reintroduced upon returning to RT. Colourless single crystals were grown upon vapour diffusion from chloroform/methanol and structure analysis confirmed pyrrole formation at each methylene bridge (Fig. 5C).
As stated above, we were unable to introduce 2- and 4-pyridyl functionality to the C[4] platform via standard reaction conditions of reflux in toluene in the presence of TsOH. Given this, we explored the use of higher boiling solvent (e.g. mesitylene), microwave syntheses, and pressurised vessels, but in every case these reactions returned starting materials. This likely relates to differences in nucleophilicity compared to 3-AP, and is unfortunate as the 4-pyridyl appended C[4] is a desirable compound for metal directed assembly,23 but work to isolate these (or structurally related compounds) will continue.
One of the key parameters controlling C[4] conformation or behaviour is lower-rim alkylation. Reintroducing the lower-rim hydroxyl groups is a desirable outcome for the new C[4]s reported here, particularly as the polyphenolic pocket can act as a binding site for a range of transition and lanthanide metal ion.24,25 With this goal in mind we have begun preliminary experiments on deprotecting the lower-rim of 4 (as it is isolable in the highest yield from all new compounds reported here). Removal of O-methyl groups at the C[4] lower-rim, including those in 1, can be achieved with relative ease using iodocyclohexane.24,26 Surprisingly, initial attempts under these conditions with 4 were unsuccessful, returning only starting material. Subsequent use of BBr3 was found to de-protect the lower-rim,27 though this gave a relatively poor yield of the target tetrahydroxy tetra-pyridazinyl C[4], 6. Reaction of 4 with 40 equiv. of BBr3 in dry DCM under N2 at RT for 24 h showed partial demethylation; this was evaluated by comparing integrals of the lower-rim methoxy 1H-NMR signals to those of the methyl groups on pyridazine moieties. Hardman et al. previously used 6.5 equiv. of BBr3 to fully deprotect a lower-rim methylated C[4] with one chloroalkyl chain at a methylene bridge,27 suggesting that the processes may be somewhat hindered. We therefore propose that the same problem may be occurring here, but to a greater extent given that all methylene bridges are functionalised with relatively bulky groups. Purification of this crude product via column chromatography, followed by NMR and mass spectroscopy, confirmed that 4 distinct products were formed. These were identified as the mono-, di- (distal and vicinal) and tri-methoxy pyridazine C[4]s. Increasing the reaction time to 48 h showed near-identical results upon workup, so alternative approaches were sought. Fantini and co-workers have utilised a two-step protocol when deprotecting a bis-calix[4]arene comprising two C[4]s tethered directly through a methylene bridge.28 This was found to avoid partially demethylated products and it is proposed that borate esters produced in the reaction are removed, thus preventing them from interfering with the demethylation. Inspired by this, compound 4 was dissolved in dry DCM, cooled to −78 °C, and 20 equiv. of BBr3 was added dropwise over 15 minutes. This was left to stir under N2 at −78 °C for 1 h before warming to RT stirring for 24 h. Analysis of the crude revealed that the major products after 24 h were tri- and di-methoxy species. The procedure was thus repeated and with a further 24 h stirring. Upon workup, the 1H-NMR spectrum showed the reaction had proceeded further, with only one methoxy signal present; this corresponded to tri-hydroxy pyridazine C[4] as a mixture with 6. Compound 6 was isolated in 5% yield via column chromatography, recrystallised from CHCl3, and structural analysis confirmed full deprotection (Fig. 6); this also shows the molecule has adopted a 1,3-alternate conformation due to H-bonding interactions, but one would expect this to revert to a cone conformer upon lower-rim deprotonation or metal ion binding in the polyphenolic pocket, though this will be the subject of future studies.
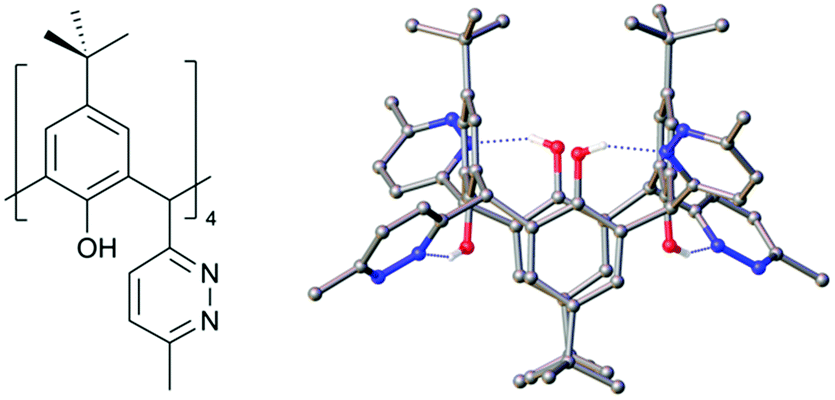 | ||
| Fig. 6 Schematic and single crystal X-ray structure of 6. Colour code: C – grey, H – white, N – blue, O – red. H atoms (except those H-bonding) omitted for clarity. | ||
In summary, we have reported new C[4] scaffolds that are monosubstituted at all methylene bridge positions with a range of functional groups. The use of a starting material in which all reactive groups are equatorial leads to retention of this arrangement in the final products, a feature that should ultimately lead to the widespread application of these molecules. Preliminary work shows that lower-rim deprotection can be achieved, though this is a priority area for optimisation. Reintroduction of key lower-rim hydroxyl functionality will allow for use of these molecules in areas such as coordination cluster formation or catalysis. Conformational changes may also lead to fascinating host–guest chemistry associated with the C[4] cavity, whilst extensive functionalisation of the appended moieties will likely lead to application in metal-directed assembly.
A. F., C. L. C. and S. H. performed experimental and analytical work. S. J. T. and S. J. D. collected XRD data. A. F., C. L. C. and S. J. D. solved the crystal structures. A. F., M. W. P. B. and S. J. D. designed the work.
We thank EPSRC for DTP support (AF/CLC). This research used resources of the Advanced Light Source, a U.S. DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.
Conflicts of interest
There are no conflicts to declare.References
- C. D. Gutsche, B. Dhawan, K. H. No and R. Muthukrishnan, J. Am. Chem. Soc., 1981, 103, 3782–3792 CrossRef CAS.
- A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713–1734 CrossRef CAS PubMed.
- J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Nature, 1994, 368, 229–231 CrossRef CAS.
- C. D. Gutsche, B. Dhawan, J. A. Levine, K. Hyun No and L. J. Bauer, Tetrahedron, 1983, 39, 409–426 CrossRef CAS.
- A. Arduini, A. Casnati, L. Dodi, A. Pochini and R. Ungaro, J. Chem. Soc., Chem. Commun., 1990, 1597–1598 RSC.
- A. Arduini, A. Pochini, A. Rizzi, A. R. Sicuri and R. Ungaro, Tetrahedron Lett., 1990, 31, 4653–4656 CrossRef CAS.
- C. D. Gutsche and L.-G. Lin, Tetrahedron, 1986, 42, 1633–1640 CrossRef CAS.
- C. D. Gutsche and L. J. Bauer, J. Am. Chem. Soc., 1985, 107, 6052–6059 CrossRef CAS.
- C. D. Gutsche, Calixarenes: An Introduction, The Royal Society of Chemistry, Cambridge, 2nd edn, 2008, ch. 5. pp. 116–146 Search PubMed.
- P. Jose and S. Menon, Bioinorg. Chem. Appl., 2007, 2007, 65815 Search PubMed.
- M. Deska, B. Dondela and W. Sliwa, ARKIVOC, 2014, 2015, 29–47 Search PubMed.
- S. E. Biali, in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer International Publishing, Switzerland, 2016, ch. 4, pp. 75–93 Search PubMed.
- C. D. Gutsche, M. Iqbal and D. Stewart, J. Org. Chem., 1986, 51, 742–745 CrossRef CAS.
- K. Agbaria and S. E. Biali, J. Am. Chem. Soc., 2001, 123, 12495–12503 CrossRef CAS PubMed.
- S. Simaan and S. E. Biali, Org. Lett., 2005, 7, 1817–1820 CrossRef CAS PubMed.
- M. Coletta, R. McLellan, P. Murphy, B. T. Leube, S. Sanz, R. Clowes, K. J. Gagnon, S. J. Teat, A. I. Cooper, M. J. Paterson, E. K. Brechin and S. J. Dalgarno, Chem. – Eur. J., 2016, 22, 8791–8795 CrossRef CAS PubMed; M. Coletta, R. McLellan, J.-M. Cols, K. J. Gagnon, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Supramol. Chem., 2016, 28, 557–566 CrossRef.
- I. Columbus and S. E. Biali, Org. Lett., 2007, 9, 2927–2929 CrossRef CAS PubMed; I. Columbus and S. E. Biali, J. Org. Chem., 2008, 73, 2598–2606 CrossRef PubMed.
- M. V. Sargent and F. M. Dean, in Comprehensive Heterocyclic Chemsitry, ed. C. W. Bird and G. W. H. Cheeseman, Pergamon, Oxford, 1984, vol. 4, pp. 599–656 Search PubMed.
- H. Hart and Y. Takehira, J. Org. Chem., 1982, 47, 4370–4372 CrossRef CAS.
- F. W. Lichtenthaler, A. Brust and E. Cuny, Green Chem., 2001, 3, 201–209 RSC.
- F. Duus, Tetrahedron, 1976, 32, 2817–2825 CrossRef CAS; M. Yuguchi, M. Tokuda and K. Orito, J. Org. Chem., 2004, 69, 908–914 CrossRef PubMed.
- V. F. Ferreira, M. C. B. V. de Souza, A. C. Cunha, L. O. R. Pereira and M. L. G. Ferreira, Org. Prep. Proced. Int., 2001, 33, 411–454 CrossRef CAS.
- P. J. Stang, D. H. Cao, S. Saito and A. M. Arif, J. Am. Chem. Soc., 1995, 117, 6273–6283 CrossRef CAS; M. Fujita, S. Nagao and K. Ogura, J. Am. Chem. Soc., 1995, 117, 1649–1650 CrossRef PubMed; Z. Zhong, A. Ikeda, M. Ayabe, S. Shinkai, S. Sakamoto and K. Yamaguchi, J. Org. Chem., 2001, 66, 1002–1008 CrossRef PubMed.
- A. Fong, L. McCormick, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Supramol. Chem., 2018, 30, 504–509 CrossRef CAS.
- M. Coletta, E. K. Brechin and S. J. Dalgarno, in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer International Publishing, Switzerland, 2016, ch. 25, pp. 671–690 Search PubMed.
- L. Zuo, S. Yao, W. Wang and W. Duan, Tetrahedron Lett., 2008, 49, 4054–4056 CrossRef CAS.
- M. J. Hardman, A. M. Thomas, L. T. Carroll, L. C. Williams, S. Parkin and J. L. Fantini, Tetrahedron, 2011, 67, 7027–7034 CrossRef CAS.
- L. T. Carroll, P. A. Hill, C. Q. Ngo, K. P. Klatt and J. L. Fantini, Tetrahedron, 2013, 69, 5002–5007 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, analytical and crystallographic information (CIFs). CCDC 2126914–2126920. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc06889j |
| This journal is © The Royal Society of Chemistry 2022 |