
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of a magnesium diboranate with organic nitriles†
Henry
Shere
,
Michael S.
Hill
 *,
Anne-Frédérique
Pécharman
and
Mary F.
Mahon
*,
Anne-Frédérique
Pécharman
and
Mary F.
Mahon
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk
First published on 11th December 2020
Abstract
A series of complexes generated through reactions of the β-diketiminato magnesium diboranate species, [(BDI)Mg{(n-Bu)pinB-Bpin}] (BDI = HC{(Me)CNDipp}2; Dipp = 2,6-di-iso-propylphenyl), and a variety of organic nitriles are reported. Although, in every case, the diboranate anion acts as a surrogate source of the {Bpin} nucleophile, resulting in B–C bond formation at the electrophilic sp-hydridised nitrile carbon, the resultant compounds display a variable propensity to undergo subsequent reaction with additional nitrile equivalents. This behaviour is rationalised to be a consequence of substituent-dependent modulation in the basicity and resultant electrophilicity of magnesium-coordinated nitrile intermediates.
Introduction
Boron compounds provide some of the most widely used main group reagents in synthetic chemistry. Their versatility is endorsed by the employment of organoborane, boronate ester and boronic acid derivatives in myriad syntheses of pharmaceuticals and other high value molecules,1–17 such as chemosensors.6,18–21 Evidence of their synthetic importance is provided by the award of two Nobel prizes for the application of boron in organic synthesis; Brown in 1979,22 for the discovery of hydroboration reactivity, and Suzuki in 2010, for the development of palladium-catalysed cross-coupling reactions to form new C–C bonds from B–C bonds.23 These processes typically involve trivalent sp2 boron reagents with a vacant p-orbital on the boron centre, such that the reactivity at boron is generally characterised by its behaviour as an electrophile.A means to induce contrasting nucleophilic character and resultant ‘umpolung’ reactivity at boron was initially identified by Schleyer, Nöth and co-workers in 1995.24,25Ab initio calculations suggested that nucleophilic singlet lithium boryls, LiBR2, may present viable synthetic targets through the introduction of stabilising electronegative R substituents (R = NH2, OH, and F). Although more than a decade passed before this prediction came to fruition, Yamashita, Nozaki and co-workers’ synthesis of the first stable boryllithium species (I, Fig. 1),26–30 by treatment of a bromoborane precursor with strongly reducing Li metal or C8K, heralded a new era in nucleophilic organoboron chemistry.
 | ||
| Fig. 1 The first boryllithium reagent, I,26 and exemplars of desymmetrised diborane (4) derivatives, II and III. | ||
Density functional theory (DFT) calculations performed on I suggested that, like Schleyer and Nöth's prototypical species, the stability of the singlet boryl unit was predicated not only on the kinetic stability provided by the bulky N-aryl substituents but also the perturbation of the frontier orbital levels induced by the π-donor and σ-acceptor characteristics of the boron-bonded nitrogen atoms.26 Although, as a further consequence, the boron atom was calculated to bear a partial positive charge, resultant nucleophilic reactivity may be considered to arise from the polarisation intrinsic to the Bδ+–Liδ++ interaction (χB: 2.04; χLi: 0.98).
Albeit the nucleophilic reactivity of I has been incontrovertibly validated through its reaction with a wide variety of organic and inorganic electrophiles,27,31–42 its synthesis necessarily requires an alkali metal reduction step to achieve the formal B(I) oxidation state. This latter feature provides a significant impediment to the wider ranging uptake of reagents like I in more conventional organic synthesis. In partial response to this issue, therefore, the subsequent revelation that a source of nucleophilic boron may be realised through the quaternisation of one of the three-coordinate boron centres of a tetra-alkoxydiborane reagent by a neutral or anionic nucleophile (e.g.II and III),43–52 represented a significant practical breakthrough in organoboron chemistry. Although monoquaternisation activates the otherwise non-polar B–B bond toward its heterolytic rupture and the conferral of apparent nucleophilic character to the remaining trigonal boron atom, such processes are most often implied from the outcome of reactions applied in situ, rather than through the explicit generation of anionic boryl intermediates.
In a manner analogous to the generation of II and III, reaction of the β-diketiminato organomagnesium derivative, [(BDI)Mgn-Bu] (IV: BDI = HC{(Me)CNDipp}2; Dipp = 2,6-di-iso-propylphenyl), with commercially available bis(pinacolato)diboron (B2pin2) results in the quaternisation of the diboron compound to yield a B(sp2)–B(sp3) diboranate species, V.53,54 Subsequent addition of strongly coordinating bases, such as 4-dimethylaminopyridine (DMAP), promotes the rupture of the B–B bond to generate magnesium boryls such as VI, through the elimination of n-BuBpin (Scheme 1).53–55 Although species such as VI are readily envisaged as sources of the {Bpin}− anion, compound V has, in its own right, been shown to act as a nucleophilic boron surrogate and as a conveniently generated reagent for nucleophilic B–C and B–B bond formation when reacted with suitable C- or B-centred electrophiles.53,56–59
 | ||
| Scheme 1 Synthesis of the magnesium diboranate, V, and the magnesium boryl, VI. | ||
Reactions of V with triphenylphosphine oxide and a variety of organic imines have implied that the mode of B–B heterolysis can encompass significant mechanistic diversity invoking either an ‘inner’ or ‘outer sphere’ approach of the Lewis basic reagent.55,56 Addition of triphenylphosphine oxide to compound V provided a Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O-coordinated magnesium boryl species, VII, which is otherwise analogous to compound VI (Scheme 2(a)). Although computational (DFT) assessment of this reaction implied that the B–B′ cleavage was induced by inner sphere coordination of the PO bond to magnesium,55 reactions of V with more sterically encumbered imine reagents provided the N–B bonded organomagnesium compounds as the initially formed kinetic products (Scheme 2(b)).56 Although these latter species may be thermally transformed into the thermodynamically-preferred amidomagnesium products of nucleophilic C-borylation, it was deduced that the process of formal boryl anion addition does not necessitate the production of any terminal Mg–B bonded intermediate. Rather, and presumably for steric reasons, initial attack of the imine nitrogen takes place at the three-coordinate boron atom of [pinB-B(n-Bu)pin]−, facilitating the effective nucleophilic displacement of charge neutral n-BuBpin.
O-coordinated magnesium boryl species, VII, which is otherwise analogous to compound VI (Scheme 2(a)). Although computational (DFT) assessment of this reaction implied that the B–B′ cleavage was induced by inner sphere coordination of the PO bond to magnesium,55 reactions of V with more sterically encumbered imine reagents provided the N–B bonded organomagnesium compounds as the initially formed kinetic products (Scheme 2(b)).56 Although these latter species may be thermally transformed into the thermodynamically-preferred amidomagnesium products of nucleophilic C-borylation, it was deduced that the process of formal boryl anion addition does not necessitate the production of any terminal Mg–B bonded intermediate. Rather, and presumably for steric reasons, initial attack of the imine nitrogen takes place at the three-coordinate boron atom of [pinB-B(n-Bu)pin]−, facilitating the effective nucleophilic displacement of charge neutral n-BuBpin.
 | ||
| Scheme 2 ‘Inner sphere’ and ‘outer sphere’ reactivity of the magnesium diboranate, V, with (a) triphenylphosphine oxide and (b) aldimines. | ||
Although B–B bond cleavage is facile in both cases, this divergent reactivity raises interesting questions with regard to the outcome of treatment of compound V with other multiply bonded organic substrates. We have earlier reported that a nuanced appreciation of reaction mechanism may be drawn from comparative variation of organic imine and nitrile substrates during their hydroboration by HBpin and catalysed by compound IV.60–62 In this contribution, therefore, we report the outcome of reactions of compound V with organic nitriles as a class of easily variable substrates presenting a similar steric profile to Ph3PO, but which, like imines, also comprise a reducible C–N multiple bond.
Results and discussion
Reactions of equimolar quantities of both iso-butyronitrile (i-PrCN) and pivalonitrile (t-BuCN) with compound V resulted in the exclusive formation of the C-borylated compounds 1 and 2, respectively (Scheme 3). The reaction with i-PrCN occurred at room temperature, while the more sterically encumbered t-BuCN required heating for 15 hours at 40 °C to induce complete conversion. In each case, two resonances were observed in the resultant 11B{1H} spectra, the chemical shifts of which were strongly reminiscent of those reported for the diboranate, V,53 (δ 34.3, 7.1 ppm) and indicative of three-coordinate and four-coordinate boron, respectively (1: δ 34.2, 8.8 ppm; 2: δ 34.4, 9.5 ppm). These data, therefore, support the proposed retention of the n-BuBpin fragment in the reaction products, in contrast to the complete elimination of n-BuBpin induced by the addition of both imine and phosphine oxide reagents (Scheme 2).55,56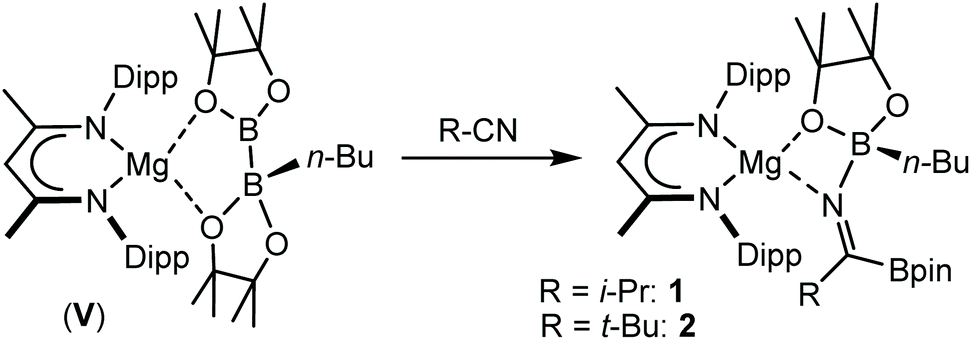 | ||
| Scheme 3 Synthesis of compounds 1 and 2. | ||
This supposition was confirmed by X-ray diffraction analysis performed on single crystals of compounds 1 and 2, which were obtained from toluene and pentane solutions, respectively (Fig. 2, Table 1). In both cases, the nitriles were revealed to have effectively inserted into the B(sp2)–B(sp3) bond of V such that the resultant C-borylated anions bind as N,O-chelating ligands to the four-coordinate Mg(1) centres via the distorted trigonal imine nitrogen and one of the pinacolatoborate oxygen atoms. The C42–N3 bond lengths of the former triple bonds of the nitrile substrates [1: 1.276(3); 2: 1.272(2) Å] are comparable to that observed within the complex anion of the previously reported C-phenylated derivative, [(BDI)Mg{PhNC(Ph)BpinPh}] [1.2967(19) Å], and indicate that the [n-Bu(Bpin)NC(R)Bpin] units of compounds 1 and 2 are similarly formulated as iminoborate anions.56 The comparable C–N, C–B and B–O bond lengths across these anions display a close correspondence, with the most notable contrast provided by the relative Z- and E-disposition of the Mg1 and (sp2) B2 atoms across the imine double bonds of 1 and 2, respectively. Despite this conformational difference, both iminoborate anions in 1 and 2 display similar N,O-coordination to magnesium via the imine nitrogen donors [Mg1–N3 (1) 2.0784(17); (2) 2.0865(14) Å] and the O1 atom of the quaternised {n-BuBpin} units [Mg1–O1 (1) 1.982(6); (2) 2.0068(12)]. Although these latter measurements are indicative of only marginally enhanced charge donation from the N-i-Pr-derived anion, it is notable that the asymmetry at B1 apparent in both solid-state structures is only reflected in the solution state NMR data of compound 1. Whereas the resonance assignable to the NC(CH3)CH protons of the BDI ligand of compound 2 appear as a single 6H singlet resonance at δ 1.63 ppm in compound 2, the equivalent environments appear as 2 differentiated 3H singlet signals in 1 (δ 1.71, 1.66 ppm).
 | ||
| Fig. 2 Molecular structures of (a) compound 1 and (b) compound 2. Hydrogen atoms, disordered atoms and solvent have been omitted for clarity. Ellipsoids are shown at 30% probability. Dipp substituents are shown as wireframe for clarity. | ||
| 1 | 2 | 4 | |
|---|---|---|---|
| a N3–C36. b B2–C36. c C36–N3–Mg1. d C36–N3–B1. | |||
| Mg1–N1 | 2.0463(17) | 2.0603(13) | 2.0483(13) |
| Mg1–N2 | 2.0640(16) | 2.0811(13) | 2.0792(14) |
| Mg1–N3 | 2.0784(17) | 2.0865(14) | 2.0899(14) |
| Mg1–O1 | 1.982(6) | 2.0068(12) | 2.0069(11) |
| N3–C42 | 1.276(3) | 1.272(2) | 1.283(2)a |
| B2–C42 | 1.584(4) | 1.600(2) | 1.600(2)b |
| N3–B1 | 1.576(3) | 1.598(2) | 1.5963(19) |
| N1–Mg1–N2 | 94.03(7) | 94.73(5) | 94.30(6) |
| N1–Mg1–N3 | 137.93(8) | 135.13(5) | 128.51(5) |
| N2–Mg1–N3 | 115.87(7) | 112.76(5) | 114.76(6) |
| O1–Mg1–N1 | 117.5(2) | 120.34(5) | 125.04(5) |
| O1–Mg1–N2 | 127.4(2) | 126.32(5) | 125.80(5) |
| O1–Mg1–N3 | 67.9(2) | 71.10(5) | 71.16(5) |
| C42–N3–Mg1 | 137.51(16) | 147.78(11) | 143.53(11)c |
| C42–N3–B1 | 130.05(19) | 122.56(13) | 127.41(13)d |
| B1–N3–Mg1 | 92.13(12) | 88.86(9) | 88.88(9) |
Although we have not investigated these processes in any further detail, our preference is to suggest that the observation of C–B(sp2) bond formation in both compounds 1 and 2 is most likely redolent of an ‘inner sphere’ mechanism reminiscent of that depicted in Scheme 2a.55 Under this regime the initial engagement of the sterically undemanding nitrile occurs at the magnesium centre of V to effect B–B heterolysis and the transient and unobservable formation of a nitrile-coordinated magnesium boryl species, which is immediately consumed by nucleophilic attack of boron at the electrophilic sp-hybridised nitrile carbon. The formation of compounds 1 and 2 may be rationalised to result from re-engagement of n-Bu-Bpin, which may or may not itself necessarily vacate the Mg coordination sphere during the reaction, with the as formed C-borylated magnesium imidate.
In an attempt to induce further reaction and displacement of the n-BuBpin components of compounds 1 and 2, the reactions were repeated in a respective 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between the relevant alkyl nitrile and compound V. Although this procedure again resulted, even after heating to 60 °C, in the consumption of one equivalent of t-BuCN and the formation of compound 2, analogous thermal treatment of the reaction with i-PrCN after the initial formation of compound 1 induced the consumption of the second nitrile equivalent to produce a predominant new compound (3) (Scheme 4).
:1 ratio between the relevant alkyl nitrile and compound V. Although this procedure again resulted, even after heating to 60 °C, in the consumption of one equivalent of t-BuCN and the formation of compound 2, analogous thermal treatment of the reaction with i-PrCN after the initial formation of compound 1 induced the consumption of the second nitrile equivalent to produce a predominant new compound (3) (Scheme 4).
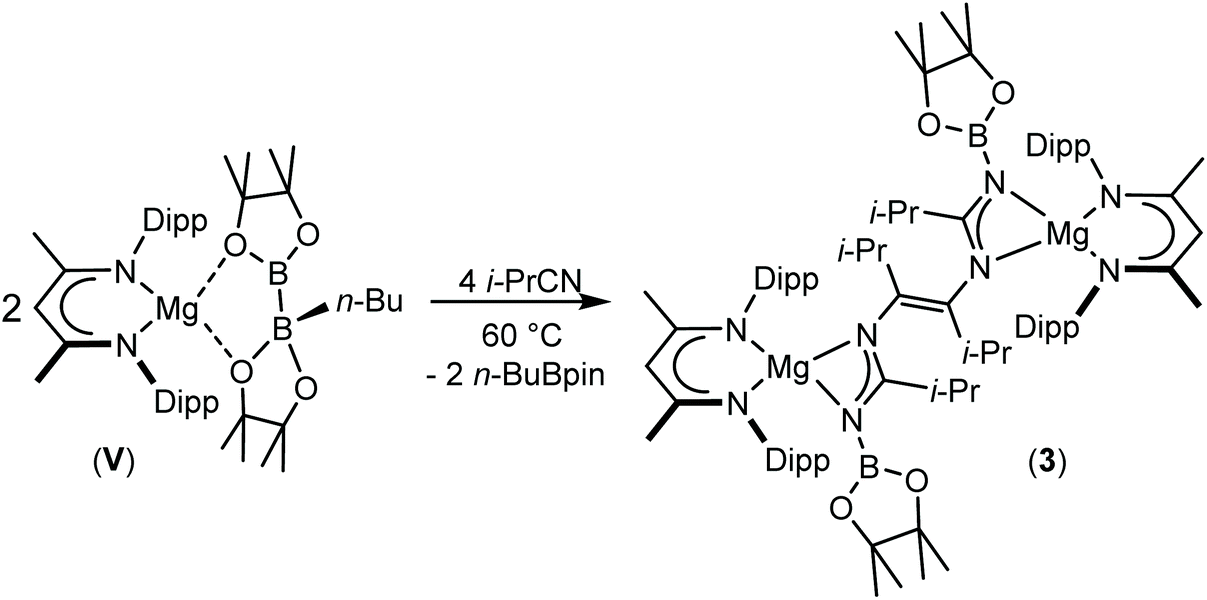 | ||
| Scheme 4 Synthesis of compound 3. | ||
Compound 3 was characterised by the appearance of a diagnostic new BDI γ-methine resonance in the resultant 1H NMR spectrum, while the corresponding 11B NMR spectrum provided evidence for the formation of uncomplexed n-BuBpin (δ 34.6 ppm), which appeared alongside a new signal at δ 10.7 ppm. The origin of these observations was resolved by slow cooling of the reaction mixture to room temperature, which induced the deposition of needle-like single crystals suitable for X-ray diffraction analysis (Fig. 3). The centrosymmetric structure of compound 3 comprises two {(BDI)Mg} units in which the magnesium centres are also coordinated by N-borylated amidinate ligands. The amidinate anions are connected with a transoid disposition across a newly formed CC double bond [C30–C30′ 1.348(2) Å]. While a number of plausible mechanistic scenarios may be envisaged, we have not studied the formation of compound 3 in detail and, thus, decline to speculate upon the precise order of events leading to the formation of compound 3. It appears, however, that addition of a further equivalent of the nitrile substrate to compound 1 results in the displacement of the coordinated equivalent of n-BuBpin and a resultant cascade of reactivity involving C-to-N migration of the remaining {Bpin} unit, C–N bond formation to the second equivalent of i-PrCN and what may be viewed as a reductive coupling of the two resultant units. In mitigation of this tentative hypothesis, the former process is reminiscent of the silyl migration reactivity exploited in Lappert and co-worker's route to a variety of silyl aza-allyl and β-diketiminate species,63,64 while there is ample precedent for CC and C–C bond formation between nitrile sp-carbon centres at a variety of low oxidation early transition metal and main group element centres.65–76
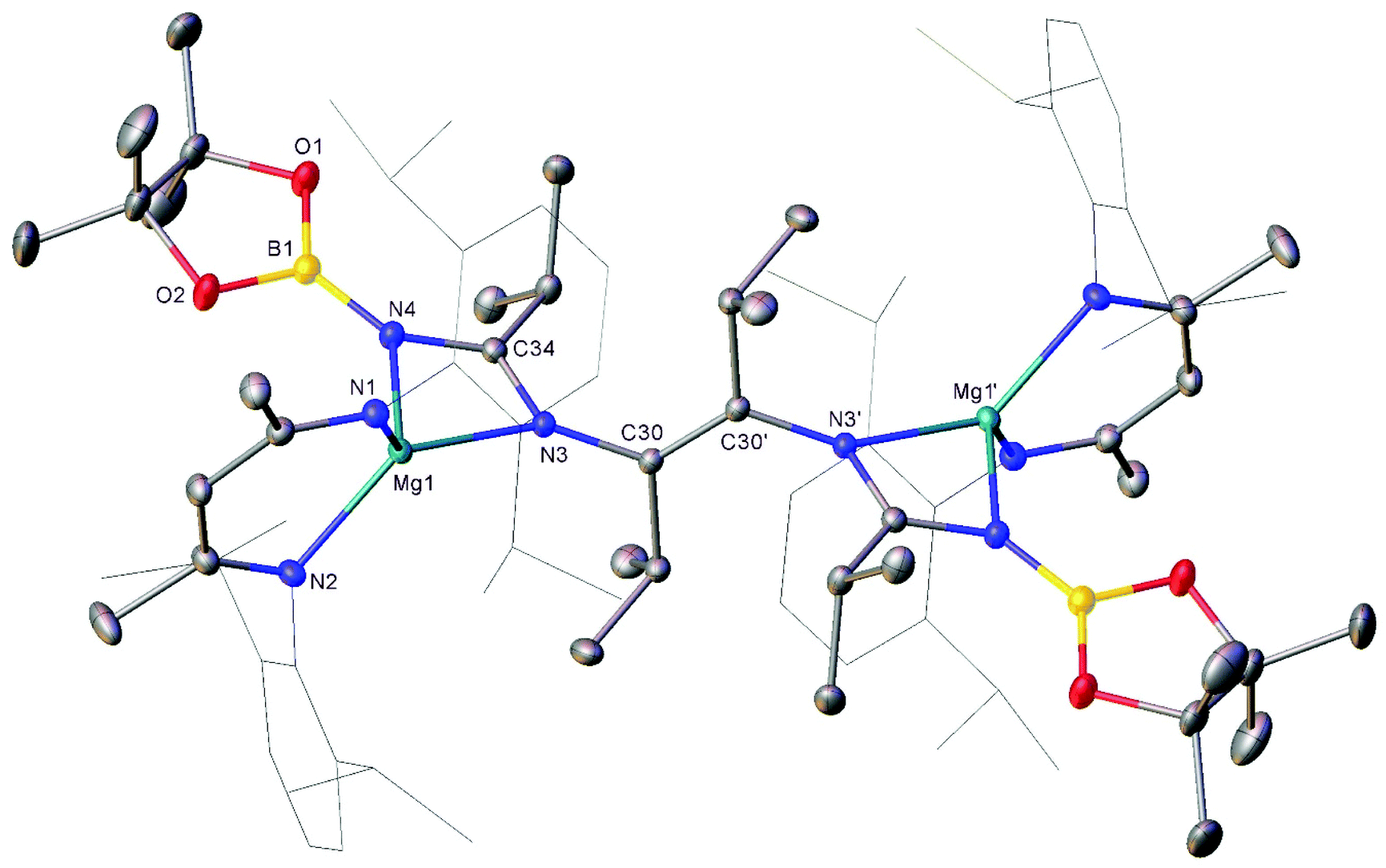 | ||
| Fig. 3 Molecular structure of compound 3. Hydrogen atoms, disordered atoms and solvent have been omitted for clarity. Ellipsoids shown at 30% probability level. Dipp substituents are shown as wireframe for clarity. Symmetry operation to generate primed atoms 1 − x, 1 − y, 1 − z. Selected bond distances (Å) and angles (°): Mg1–N1 2.0501(10), Mg1–N2 2.0536(10), Mg1–N3 2.1343(9), Mg1–N4 2.0445(10), N3–C30 1.4385(13), N3–C34 1.3265(14), N4–C34 1.3509(14), N4–B1 1.4102(16), C30–C30′ 1.348(2), N1–Mg1–N2 93.25(4), N1–Mg1–N3 125.92(4), N2–Mg1–N3 133.02(4), N4–Mg1–N1 120.61(4), N4–Mg1–N2 120.39(4), N4–Mg1–N3 64.62(4). | ||
The presence of its potentially diagnostic methyl group resonance in resultant NMR spectra led us to select o-tolunitrile as the preferred reagent to extend this study to aryl-substituted nitriles. Although examination of the 1H NMR spectrum recorded immediately after addition of a stoichiometric quantity of o-tolunitrile to compound V at room temperature indicated the formation of a mixture of compounds, the anticipated product, compound 4, could be isolated in 41% yield as colourless single crystals after fractional crystallisation of the reaction mixture (Scheme 5). The resultant X-ray analysis (Fig. 4(a), Table 1) revealed that 4 is structurally analogous to compounds 1 and 2.
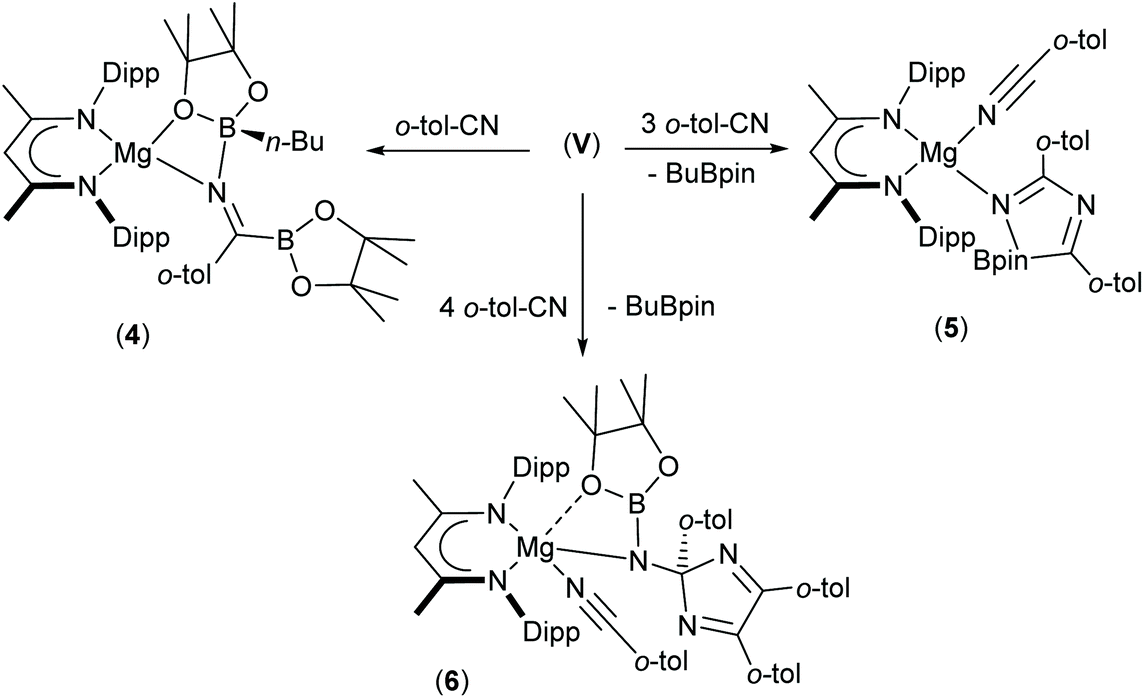 | ||
| Scheme 5 Synthesis of compounds 4–6. | ||
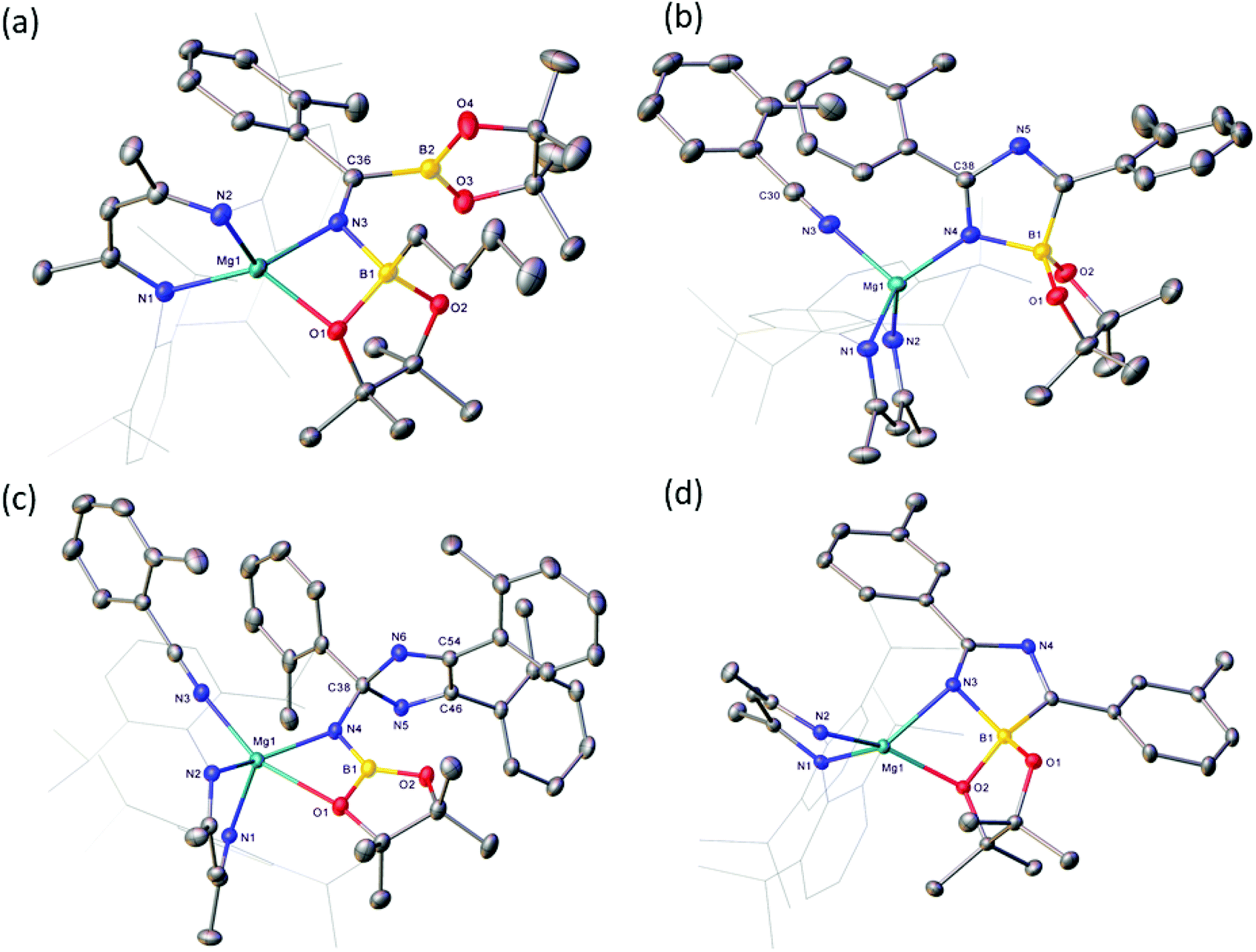 | ||
| Fig. 4 Molecular structures of (a) compound 4, (b) compound 5, (c) compound 6 and (d) compound 7. Hydrogen atoms and disordered atoms and have been omitted for clarity, as has the solvent present in 4, 5 and 6. Ellipsoids are depicted at 30% probability. Dipp substituents are shown as wireframe for clarity. | ||
Inspection of the metric data presented in Table 1 indicates that the replacement of the iso-propyl and tert-butyl groups of 1 and 2 by ortho-tolyl exerts negligible impact on the nature of the resultant iminoborate anion in 4. The less discriminating outcome of the equimolar reaction leading to the formation of compound 4, however, led us to further assess the reactions of V with greater stoichiometric equivalents of o-tolunitrile (Scheme 5). Analysis by 1H NMR spectroscopy immediately after reaction in a 1:2 ratio at room temperature indicated the generation of significant quantities of a new species, compound 5, which was most clearly evidenced by the appearance of a BDI γ-methine signal at δ 5.04 ppm. This product also predominated in subsequent reactions performed at room temperature with 3 equivalents of o-tolunitrile. In contrast, analysis of a reaction between V and a fourfold excess of the nitrile allowed the identification of a further minor compound (6), manifested as an additional methine singlet resonance at δ 4.91 ppm. The formation of both compounds ensued with simultaneous elimination of n-BuBpin, while heating of the latter reaction to 60 °C resulted in the complete transformation of compound 5 to compound 6. The basis of these observations was resolved by X-ray diffraction analysis of both compounds 5 and 6 after crystallisation from hexane solution (Scheme 5). The structures of both compounds are illustrated as Fig. 4(b) and (c), respectively, while selected bond length and angle data are presented in Table 2. The 4-coordinate β-diketiminato magnesium centre of compound 5 is further ligated by the nitrogen atom of a molecule of o-tolunitrile [Mg1–N3 2.138(2) Å] and the nitrogen centre [Mg1–N4 2.068(2) Å] of a spirocyclic borate anion in which the distorted tetrahedral boron geometry is provided by a O,O′-bidentate pinacolate unit and a 5-membered C,N-chelate. The alternating N4–C38 [1.2822(3) Å], C38–N5 [1.449(3) Å] and N5–C46 [1.288(3) Å] bond lengths of this latter heterocycle indicate that it is best viewed as a 1-bora-2,4-diazabutadiene formed by the incorporation of two molecules of o-tolunitrile. As mentioned previously, nitrile–nitrile C–C dimerisation leading to metalla-1,4-diaza-butadiene complexes is a quite well precedented process.65–76 To the best of our knowledge, however, the only previous report of nitrile C–N coupling to yield a related 2,4-diazabutadiene derivative appears to be provided by a serendipitous iron-centred dimerisation of cyanamide.77 The [(BDI)Mg] unit of compound 6 is similarly coordinated by a molecule of o-tolunitrile [Mg1–N3 2.1607(13) Å] and a bidentate borylamide ligand [Mg1–N4 2.0288(11) Å], which forms a 4-membered chelate via an additional donor interaction with a pinacolate oxygen atom [Mg1–O1 2.5318(11) Å] and raises the magnesium coordination number to five.62,78 The N4 atom of the borylamide anion is also bonded to a 2,4,5-tri-o-tolyl-2H-imidazole unit [N4–C38 1.4442(16) Å] such that the overall constitution of the coordinated amide ligand of 6 may be rationalised as resulting from the addition of a further equivalent of the nitrile reagent to the borate anion observed in 5.
| 5 | 6 | 7 | |
|---|---|---|---|
| a Mg1–O2. b N3–C30. c N4–C30. d N4–C38. e B1–N3. f B1–C38. g N3–Mg1–O2. | |||
| Mg1–N1 | 2.0466(18) | 2.0860(11) | 2.0260(10) |
| Mg1–N2 | 2.0250(18) | 2.0676(11) | 2.0556(10) |
| Mg1–N3 | 2.138(2) | 2.1607(13) | 2.0502(11) |
| Mg1–N4 | 2.068(2) | 2.0288(11) | — |
| Mg1–O1 | — | 2.5318(11) | 2.0360(9)a |
| N4–C38 | 1.282(3) | 1.4442(16) | 1.2815(16)b |
| N5–C38 | 1.449(3) | 1.4927(16) | 1.4359(15)c |
| N5–C46 | 1.288(3) | 1.2762(18) | 1.2981(16)d |
| N6–C38 | — | 1.4972(17) | — |
| N6–C54 | — | 1.2758(18) | — |
| C46–C54 | — | 1.5041(18) | — |
| B1–N4 | 1.633(3) | 1.3975(19) | 1.5961(16)e |
| B1–C46 | 1.662(3) | — | 1.6681(16)f |
| N1–Mg1–N2 | 94.21(7) | 92.48(5) | 94.69(4) |
| N1–Mg1–N3 | 115.72(8) | 109.28(5) | 129.87(4) |
| N1–Mg1–N4 | 114.49(8) | 119.65(5) | — |
| N2–Mg1–N3 | 93.63(8) | 92.26(5) | 112.77(4) |
| N2–Mg1–N4 | 127.66(8) | 135.01(5) | — |
| N4–Mg1–O1 | — | 61.85(4) | 69.63(4)g |
We have previously observed that the mechanisms leading to the magnesium-catalysed dihydroboration of organic nitriles with HBpin display a subtle dependence on the identity of the organic substrate.62 Although proceeding via a common pathway, the rate determining processes were dependent on the establishment of several pre-equilibria, the positions of which were dictated by the N(sp)-basicity and C(sp)-electropilicity of the nitrile function, with particularly contrasting behaviour observed for aliphatic versus aromatic substitution. It is well known that coordination of nitriles to transition metal ions results in an enhanced electrophilicity of the nitrile carbon, rendering it more susceptible to nucleophilic attack.79 Similarly, the kinetic facility of the base hydrolysis of coordinated aryl nitriles displays a sensitive dependence on Hammett σ substituent constants.80 On this basis, therefore, we suggest that the contrasting stability of the initially formed aryl iminoborate anion of compound 4 with those of compounds 1 and 2 toward onward reaction with further nitrile equivalents is primarily a consequence of similar electronic adjustments dictated by coordination of the nitrile to magnesium. In this case, the reactivity of compound 4 toward additional nitrile equivalents may be attributed to the enhanced electrophilicity of the sp-hybridised nitrile carbon centre of o-tolunitrile. We suggest, therefore, that the formation of compounds 5 and 6 derives from the sequential coordination and reaction of o-tolunitrile as depicted in Scheme 6. Although the transformation of compound 4 is driven by the basicity of o-tolunitrile and its ability to displace n-BuBpin, its coordination to magnesium renders the aryl nitrile more susceptible to intramolecular nucleophilic attack. The forward trajectory of the reaction and the maintenance of the borate species (5) is, thus, facilitated by the necessary coordination of a further equivalent of nitrile to magnesium. The transformation of compound 5 to compound 6 is then envisaged in an effectively identical fashion, such that the reaction sequence may be rationalised as a consequence of the nitrogen-centered basicity and coordinative ability of the ortho-methyl substituted aryl nitrile alongside a resultant enhancement in its carbon-centred electrophilicity.
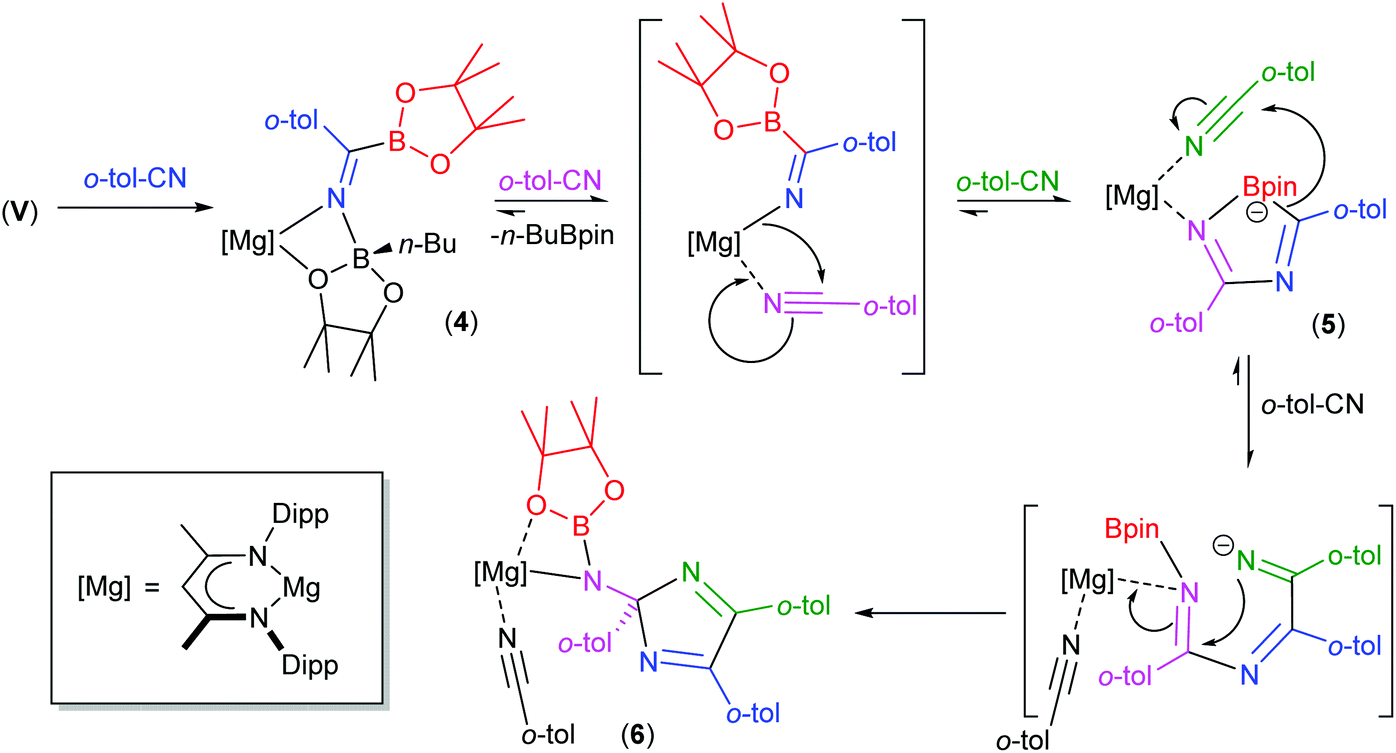 | ||
| Scheme 6 Suggested sequential addition of o-tolunitrile to compound V and the formation of compounds 5 and 6. | ||
The plausibility of this mechanistic hypothesis was borne out by a further reaction performed between compound V and the less basic m-tolunitrile. Analysis of a reaction in a 2:1 stoichiometry by 1H NMR spectroscopy after heating at 60 °C evidenced the formation of a major new compound (7), which was characterised by a BDI γ-methine C–H resonance at δ 5.02 ppm. Removal of volatiles and crystallisation from hexane provided pale yellow crystals suitable for X-ray diffraction analysis. The results of this analysis (Fig. 4(d), Table 2) identified compound 7 as a further borate species analogous to compound 5. Although the spirocyclic anion again engages via a formal imino nitrogen atom of the 1-bora-2,4-diazabutadiene component [Mg1–N3 2.0502(11) Å], the anion interacts as a four-membered chelate toward the magnesium centre via a further short contact with an oxygen atom of the pinacolate unit [Mg1–O2 2.0360(9) Å]. The robust nature of this interaction was underscored by further reactions performed between compound V and three and four stoichiometric equivalents of m-tolunitrile, which also yielded compound 7 as the predominant reaction product. Although we cannot completely discount the influence of the differing steric profiles of the two nitrile substrates, we suggest that the borate anion comprising compound 7 forms in an analogous fashion to that deduced for compound 5. The lower basicity induced by the inductive effect of meta-substitution, however, perturbs the further necessary nitrile coordination and the formation of a m-tolyl variant of compound 7.
Conclusion
A series of complexes generated through reactions of a magnesium diboranate species and a variety of organic nitriles has been prepared. Although in each case the diboranate anion acts as a surrogate source of the {Bpin} nucleophile, through addition to the electrophilic sp-hydridised nitrile carbon, the resultant compounds display a variable propensity to undergo subsequent reaction with additional nitrile equivalents. This behaviour is rationalised to be a consequence of substituent-dependent variations in the basicity and resultant electrophilicity of the magnesium-coordinated nitrile intermediates. We are continuing to study the reactivity of similar magnesium compounds with various electrophiles and are seeking to extend these protocols to the heavier elements of the alkaline earth series.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (EP/N014456/1 and EP/R020752/1) and the University of Bath for the provision of a PhD scholarship (HS).References
- W. Q. Yang, X. M. Gao and B. H. Wang, Med. Res. Rev., 2003, 23, 346–368 CrossRef CAS.
- S. J. Baker, C. Z. Ding, T. Akama, Y. K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS.
- P. C. Trippier and C. McGuigan, MedChemComm, 2010, 1, 183–198 RSC.
- S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 RSC.
- S. Touchet, F. Carreaux, B. Carboni, A. Bouillon and J. L. Boucher, Chem. Soc. Rev., 2011, 40, 3895–3914 RSC.
- J. S. Hansen, J. B. Christensen, J. F. Petersen, T. Hoeg-Jensen and J. C. Norrild, Sens. Actuators, B, 2012, 161, 45–79 CrossRef CAS.
- R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156–4220 CrossRef CAS.
- B. C. Das, P. Thapa, R. Karki, C. Schinke, S. Das, S. Kambhampati, S. K. Banerjee, P. Van Veldhuizen, A. Verma, L. M. Weiss and T. Evans, Future Med. Chem., 2013, 5, 653–676 CrossRef CAS.
- H. Fu, H. Fang, J. Sun, H. Wang, A. Liu and Z. Wu, Curr. Med. Chem., 2014, 21, 3271–3280 CrossRef CAS.
- Z. J. Lesnikowski, Expert Opin. Drug Discovery, 2016, 11, 569–578 CrossRef CAS.
- T. B. Clark, Asian J. Org. Chem., 2016, 5, 31–42 CrossRef CAS.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS.
- F. K. Scharnagl, S. K. Bose and T. B. Marder, Org. Biomol. Chem., 2017, 15, 1738–1752 RSC.
- S. Thareja, M. Y. Zhu, X. Y. Ji and B. H. Wang, Heterocycl. Commun., 2017, 23, 137–153 CAS.
- F. Yang, M. Y. Zhu, J. Y. Zhang and H. C. Zhou, MedChemComm, 2018, 9, 201–211 RSC.
- J. P. M. Antonio, R. Russo, C. P. Carvalho, P. Cal and P. M. P. Gois, Chem. Soc. Rev., 2019, 48, 3513–3536 RSC.
- G. F. S. Fernandes, W. A. Denny and J. L. Dos Santos, Eur. J. Med. Chem., 2019, 179, 791–804 CrossRef CAS.
- S. Huang, M. Jia, Y. Xie, J. Wang, W. Xu and H. Fang, Curr. Med. Chem., 2012, 19, 2621–2637 CrossRef CAS.
- J. S. Hansen and J. B. Christensen, Biosensors, 2013, 3, 400–418 CrossRef CAS.
- E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831–3842 RSC.
- X. L. Sun and T. D. James, Chem. Rev., 2015, 115, 8001–8037 CrossRef CAS.
- H. C. Brown and S. Krishnamurthy, Tetrahedron, 1979, 35, 567–607 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- M. Wagner, N. Hommes, H. Nöth and P. V. Schleyer, Inorg. Chem., 1995, 34, 607–614 CrossRef CAS.
- H. M. Tuononen, R. Roesler, J. L. Dutton and P. J. Ragogna, Inorg. Chem., 2007, 46, 10693–10706 CrossRef CAS.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS.
- M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570–9571 CrossRef CAS.
- Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS.
- M. Yamashita and K. Nozaki, J. Synth. Org. Chem., Jpn., 2010, 68, 359–369 CrossRef CAS.
- M. Yamashita and K. Nozaki, in Synthesis and Application of Organoboron Compounds, ed. E. Fernandez and A. Whiting, 2015, vol. 49, pp. 1–37 Search PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef CAS.
- T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162–14163 CrossRef CAS.
- K. Nozaki, Y. Aramaki, M. Yamashita, S. H. Ueng, M. Malacria, E. Lacote and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 11449–11451 CrossRef CAS.
- T. Arnold, H. Braunschweig, W. C. Ewing, T. Kramer, J. Mies and J. K. Schuster, Chem. Commun., 2015, 51, 737–740 RSC.
- L. M. A. Saleh, K. H. Birjkumar, A. V. Protchenko, A. D. Schwarz, S. Aldridge, C. Jones, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2011, 133, 3836–3839 CrossRef CAS.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS.
- A. V. Protchenko, D. Dange, J. R. Harmer, C. Y. Tang, A. D. Schwarz, M. J. Kelly, N. Phillips, R. Tirfoin, K. H. Birjkumar, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Nat. Chem., 2014, 6, 315–319 CrossRef CAS.
- J. Campos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 14159–14163 CrossRef CAS.
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128–7131 RSC.
- R. Frank, J. Howell, J. Campos, R. Tirfoin, N. Phillips, S. Zahn, D. M. P. Mingos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 9586–9590 CrossRef CAS.
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS.
- M. S. Cheung, T. B. Marder and Z. Y. Lin, Organometallics, 2011, 30, 3018–3028 CrossRef CAS.
- K. S. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253–7254 CrossRef CAS.
- C. Kleeberg, A. G. Crawford, A. S. Batsanov, P. Hodgkinson, D. C. Apperley, M. S. Cheung, Z. Y. Lin and T. B. Marder, J. Org. Chem., 2012, 77, 785–789 CrossRef CAS.
- S. K. Bose, K. Fucke, L. Liu, P. G. Steel and T. B. Marder, Angew. Chem., Int. Ed., 2014, 53, 1799–1803 CrossRef CAS.
- R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC.
- S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Y. Mo, D. Qiu, M. S. Cheung, L. Dang, J. B. Wang, U. Radius, Z. Y. Lin, C. Kleeberg and T. B. Marder, Chem. – Eur. J., 2015, 21, 7082–7098 CrossRef CAS.
- A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyas and E. Fernandez, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 CrossRef CAS.
- H. Gulyas, A. Bonet, C. Pubill-Ulldemolins, C. Sole, J. Cid and E. Fernandez, Pure Appl. Chem., 2012, 84, 2219–2231 CAS.
- C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyas and E. Fernandez, Chem. – Eur. J., 2012, 18, 1121–1126 CrossRef CAS.
- N. Miralles, J. Cid, A. B. Cuenca, J. J. Carbo and E. Fernandez, Chem. Commun., 2015, 51, 1693–1696 RSC.
- A. B. Cuenca, R. Shishido, H. Ito and E. Fernandez, Chem. Soc. Rev., 2017, 46, 415–430 RSC.
- A. F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef.
- A. F. Pécharman, M. S. Hill and M. F. Mahon, Dalton Trans., 2018, 47, 7300–7305 RSC.
- A. F. Pécharman, N. A. Rajabi, M. S. Hill, C. L. McMullin and M. F. Mahon, Chem. Commun., 2019, 55, 4 RSC.
- A. F. Pécharman, M. S. Hill, G. McMullon, C. L. McMullin and M. F. Mahon, Chem. Sci., 2019, 10, 6672–6682 RSC.
- A. F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Angew. Chem., Int. Ed., 2017, 56, 16363–16366 CrossRef.
- A. F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Organometallics, 2018, 37, 4457–4464 CrossRef.
- A. F. Pécharman, M. S. Hill and M. F. Mahon, Angew. Chem., Int. Ed., 2018, 57, 10688–10691 CrossRef.
- M. Arrowsmith, M. S. Hill and G. Kociok-Koehn, Chem. – Eur. J., 2013, 19, 2776–2783 CrossRef CAS.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem. Commun., 2015, 51, 14477–14480 RSC.
- C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628–641 RSC.
- P. B. Hitchcock, M. F. Lappert and D.-S. Liu, J. Chem. Soc., Chem. Commun., 1994, 1699–1700 RSC.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS.
- M. Ma, A. Stasch and C. Jones, Chem. – Eur. J., 2012, 18, 10669–10676 CrossRef CAS.
- M. G. Gardiner, A. N. James, C. Jones and C. Schulten, Dalton Trans., 2010, 39, 6864–6870 RSC.
- G. Tomaschun, K. Altenburger, F. Reiß, L. Becker, A. Spannenberg, P. Arndt, H. Jiao and U. Rosenthal, Eur. J. Inorg. Chem., 2016, 272–280 CrossRef CAS.
- G. R. Kiel, A. E. Samkian, A. Nicolay, R. J. Witzke and T. D. Tilley, J. Am. Chem. Soc., 2018, 140, 2450–2454 CrossRef CAS.
- L. Becker, P. Arndt, A. Spannenberg and U. Rosenthal, Chem. – Eur. J., 2014, 20, 12595–12600 CrossRef CAS.
- L. Becker, F. Reiss, K. Altenburger, A. Spannenberg, P. Arndt, H. Jiao and U. Rosenthal, Chem. – Eur. J., 2016, 22, 10826–10838 CrossRef CAS.
- L. Becker, P. Arndt, H. Jiao, A. Spannenberg and U. Rosenthal, Angew. Chem., Int. Ed., 2013, 52, 11396–11400 CrossRef CAS.
- L. Becker, P. Arndt, A. Spannenberg, H. Jiao and U. Rosenthal, Angew. Chem., Int. Ed., 2015, 54, 5523–5526 CrossRef CAS.
- L. Becker, F. Strehler, M. Korb, P. Arndt, A. Spannenberg, W. Baumann, H. Lang and U. Rosenthal, Chem. – Eur. J., 2014, 20, 3061–3068 CrossRef CAS.
- L. Becker, M. Haehnel, A. Spannenberg, P. Arndt and U. Rosenthal, Chem. – Eur. J., 2015, 21, 3242–3248 CrossRef CAS.
- Q. X. Dai, H. Seino and Y. Mizobe, Organometallics, 2014, 33, 3652–3655 CrossRef CAS.
- L. Zhang, B. Fang, G. Hou, L. Ai, W. Ding, M. D. Walter and G. Zi, Dalton Trans., 2016, 45, 16441–16452 RSC.
- N. A. Spahn, M. H. Nguyen, J. Renner, T. K. Lane and J. Louis, J. Org. Chem., 2017, 82, 234–242 CrossRef CAS.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and A. S. S. Wilson, Angew. Chem., Int. Ed., 2015, 54, 13362–13365 CrossRef CAS.
- R. A. Michelin, M. Mozzon and R. Bertani, Coord. Chem. Rev., 1996, 147, 299–338 CrossRef CAS.
- R. L. De La Vega, W. R. Ellis Jr. and W. L. Purcell, Inorg. Chim. Acta, 1983, 68, 97–101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details NMR spectra and details of the crystallographic analysis of compounds 1–7. CCDC 2044859–2044865 for compounds 1–7, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt04016a |
| This journal is © The Royal Society of Chemistry 2021 |