
A perspective on practical solar to carbon monoxide production devices with economic evaluation†
Sang Youn
Chae
 ab,
Si Young
Lee
ac,
Sung Gyu
Han
a,
Honggon
Kim
a,
Jongwon
Ko
d,
Sejin
Park
d,
Oh-Shim
Joo
a,
Donghwan
Kim
ce,
Yoonmook
Kang
e,
Ung
Lee
*ace,
Yun Jeong
Hwang
*ac and
Byoung Koun
Min
*ae
ab,
Si Young
Lee
ac,
Sung Gyu
Han
a,
Honggon
Kim
a,
Jongwon
Ko
d,
Sejin
Park
d,
Oh-Shim
Joo
a,
Donghwan
Kim
ce,
Yoonmook
Kang
e,
Ung
Lee
*ace,
Yun Jeong
Hwang
*ac and
Byoung Koun
Min
*ae
aClean Energy Research Center, Korea Institute of Science and Technology, Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: ulee@kist.re.kr; yjhwang@kist.re.kr; bkmin@kist.re.kr
bDepartment of Materials Science, Institute for Surface Science and Corrosion WW4-LKO, University of Erlangen-Nuremberg, Martensstrasse 7, 91058 Erlangen, Germany
cDivision of Energy and Environmental Technology, KIST School, Korea University of Science and Technology, Seoul 02792, Republic of Korea
dDepartment of Materials Science and Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
eGreen School, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
First published on 14th October 2019
Abstract
Solar-chemical production is one of the most promising options for producing valuable chemicals from greenhouse gases. An economically attractive and industrially applicable solar-chemical production device not only requires catalyst and/or reactor design, but also auxiliary unit process design, process integration, and optimization. Herein, we report a state-of-the-art monolithic solar-chemical production device having 8.03% solar to CO conversion efficiency and 0.77 to 31.9% CO2 one path conversion. Since the monolithic device directly couples a photovoltaic cell and a CO2 electrolyzer, the power loss due to a current converter can be avoided. According to the solar-chemical production device, a comprehensive process design accounting for CO2 to CO conversion, unreacted CO2 separation, and recycling structure is provided. The process model shows good agreement with experimental data for CO2 conversion in the electrolyzer. A process level techno-economic evaluation and a comprehensive review are also presented to highlight the current state and the economic feasibility of the developed device. Thereafter, we provide a sensitivity analysis in terms of CO2 conversion, membrane cost, solar to chemical efficiency, and current density necessary for economically profitable CO production. The equivalent CO sales cost from a 4 MW production plant is estimated to be $10.9 per kg and the corresponding carbon tax compensating for the price gap of the current market price is $6.6 per kg CO2. The sensitivity analysis demonstrates that >80 mA cm−2 current density or 22% CO2 conversion is desirable to effectively compete with the conventional CO production process.
Introduction
Global dependence on fossil fuels for energy and chemical precursors makes them an irreplaceable resource. Indiscriminate consumption, however, will stunt the development of human society. The time scale for the formation of fossil fuels is several million years; thus known fossil fuel supplies will not be able to meet the exponentially growing demand in the near future.1,2 In response to the imminent fuel shortage, much emphasis has been placed on producing electricity from renewable energy sources such as sunlight. However, electricity production does not take into account the need for carbon-containing precursors for plastics, pharmaceuticals, etc. Therefore, artificial photosynthetic solar-chemical production that converts CO2 into fuels and chemical precursors is proposed as a solution for replacing fossil fuels.3Solar-chemical production can be achieved using three systems: photocatalytic (PC), photoelectrochemical cell (PEC), and photovoltaic driven electrochemical cell (PV–EC) systems. Although their configurations vary, they share a similar concept of harvesting light to form electron–hole pairs that facilitate redox chemistry. In the PC system, both the spontaneous electron–hole pair formation and the redox reaction occur within semiconductor materials. The major challenge with this system is preventing the recombination of the photo-generated charge carriers before chemical production. Common approaches to overcome it include eliminating defective recombination centers or introducing co-catalysts to increase reaction kinetics. However, PC systems still have poor quantum efficiency and thus low solar to chemical energy conversion efficiency in comparison with others, even though heating water and/or adding a sacrificial agent may improve the quantum efficiency of PC systems.4–6
Alternatively, oxidation and reduction reactions can respectively occur at the anode and the cathode of the PEC system by separating hole- and electron-involved reactions in order to increase charge separation at the semiconductor/liquid junction. In addition, insufficient electronic band positions or photovoltages of the semiconductor photoelectrode can be adjusted by the external bias potential to initiate catalytic chemical reactions. The tandem structure of multi-junction semiconductors has been demonstrated to increase the light absorption efficiency as well as solar to chemical conversion efficiency in water splitting or CO2 reduction, but the complicated structures with limited conversion efficiency are not favorable from a practical point of view.11 Moreover, the photocorrosion of the semiconductor material is still of concern for long-term operation when the photoelectrode comes into contact with the electrolyte under illumination and/or under biased potentials.
On the other hand, a PV–EC system can decouple the light absorption and electrochemical (EC) reaction processes. The electric driving force provided by photovoltaics (PV) allows great expansion of the electrode material choice, as materials are no longer restricted to being semiconductors. Currently, the PV–EC system is the most promising technology for practical applications owing to its high solar to chemical conversion efficiency and technical maturity for large scale production processes.10,12
As seen in Table 1, the PV–EC system has achieved the highest solar to chemical efficiency for CO2 reduction.10 High efficiency of the PV–EC system can be achieved through its device structure because the light absorber and electrochemical catalyst are separated; therefore, it provides various options for coupling light management and the catalytic reaction. In addition, the PV photoelectrode/EC reactor integration can minimize the energy loss penalties that occur in all energy transfer processes.
| Approach | Photocatalyst/photoelectrode/photovoltaics | Co-catalyst or counter electrode | STC efficiency |
|---|---|---|---|
| PC | TiO2/InP | Metal complex | 0.04% (HCOO-)5 |
| PEC | Photocathode: CuFeO2 | Anode: Pt | 1% (HCOO-)7 |
| Photocathode: GaAs/InGaP/TiO2/Ni | Cathode: Pd/C/Ti | 10% (HCOO-)8 | |
| PV–EC | Silicon-heterojunction solar cell (series connection module) | Cathode: Cu–Ag | 5.6%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 (hydrocarbon and oxygenate) 9 (hydrocarbon and oxygenate) |
| Anode: IrO2 | |||
| 28.5% GaInP/GaInAs/Ge solar cell (triple junction tandem cell) | Cathode: CuO/SnO | 13.4% (CO)10 | |
| Anode: CuO/SnO2 |
However, the PV–EC system still suffers from the high overpotentials associated with electrochemical CO2 reduction/water oxidation, and product selectivity loss induced by the competing hydrogen evolution reaction (HER). Especially, the overpotential of the CO2 reduction is dramatically increased at high current density compared with HER due to its low solubility and slow kinetics,13 and thus the product selectivity loss can be more problematic. In addition, IR (voltage) drop induced by the series resistance of the electrolyte or device components can cause another critical energy loss as the operating current increases.
To optimize the solar to chemical conversion efficiency (ηSTF) of an artificial photosynthesis system, the driving force (photovoltage) and the production rate (current density of the reaction) must be controlled as the operating current density (Jop) when the electrochemical reactor and photovoltaic photoelectrode are coupled. Because the photovoltage (i.e., Voc) and short circuit current density (Jsc) of a PV cell involve a trade-off when its power conversion efficiency is fixed, the optimum combination of Voc and Jsc is needed to identify the most efficient EC reactor operations under the consideration of the configuration between the PV cell and the EC electrode.
In addition, the performance of electrochemical electrodes is generally diagnosed in a small-scale device using a three-electrode system, but the efficiency of the EC reactor in a real device can be affected by solution resistance, size, conversion rate, design of the PV–EC system, etc. which are not related to the catalyst properties. Moreover, the configuration of PV and EC cells has an impact not only on the solar to chemical conversion efficiency but also on the total investment cost affecting techno-economics. In fact, the EC reactor is the most expensive operation unit in the CO production processes through the PV–EC artificial photosynthesis system. The type of catalyst electrode/PV cell also has a large impact on both chemical conversion efficiency and cost. Various solar cell technologies are used for CO production or similar products (e.g., acetic acid, formate, or H2), such as perovskite solar cells, dye-sensitized solar cells, and III–V solar cells.10,12,14,15
Most PV–EC devices are coupled with a tandem solar cell aiming at achieving a high photovoltage.16,17 Despite the high power conversion efficiency obtained from the tandem structure, the fabrication process is often complicated and expensive. In addition, tandem solar cell technology is mainly concentrated on III–V group materials,18 which are difficult to use in large-scale production. The silicon solar cell is one of the best candidates for large scale solar-chemical production because of its high-power conversion efficiency, good stability, technical maturity, and large-area production. However, the single junction of a silicon solar cell can generate a Voc of only 0.6–0.7 V, which does not meet the requirements for CO2 reduction. Therefore, the module design may require series connections of silicon solar cells to supply a sufficient photovoltage for solar-chemical production.
Apart from the aforementioned challenges, the practical application of the PV–EC device should also consider a complete process design and the process economy which have been barely considered in previous photoelectrochemical CO2 reduction studies. For example, unreacted CO2 separation and the recycling process should be carefully examined and optimally selected. Also, the optimal process operation conditions need to be identified because both the process efficiency and the economic feasibility are mainly dependent on them. The electrochemical CO2 reduction process design and its economic potential can also be determined by selection of target products, reactor design, catalysts, and electrolytes and thus the optimum selection of these variables is desirable.27,28
Techno-economic analysis of solar-chemical production devices can demonstrate the economic feasibility and provide guidelines for further technical development. In fact, techno economic analysis of solar-chemical production systems has been presented in various studies. However, approximated assessments are generally conducted using different evaluation metrics; thus, it is difficult to compare different technologies in a fair manner. Estimating the cost of electrochemical devices and designing rigorous product purification processes are major challenges for building an economic model. Table 2 summarizes the most popular techno-economic assessments. Electrochemical hydrogen production is the most intensively studied process. The analyses in Table 2 suggest system selection among available options (e.g., PEC, PV–EC) and present comprehensive life cycle assessments.
| Product | Year | Economic Metrics |
|---|---|---|
| Formic acid | 2011 | Value chain analysis |
| Life cycle net energy assessment19 | ||
| Hydrogen | 2014 | Using the energy return on energy invested (EROEI) and the energy payback time (EPBT)20 |
| Methanol | 2015 | The energy incorporation efficiency (EIE)21 |
| Syngas/Fischer–Tropsch | 2016 | The levelized cost22 |
| Formic acid | 2016 | Gross margin, benefit cost ratio (BCR) net present value (NPV)22 |
| Hydrogen | 2016 | The levelized cost using NPV23 |
| Hydrogen, formic acid, carbon monoxide, methanol, methane, ethylene, ethanol, carbon monoxide, syngas | 2016 | Gross margin24 |
| Formic acid, methanol, methane | 2018 | NPV25 |
| Ethylene, ethanol, n-propanol, various commodity chemicals | 2018 | Modified Douglas “level 2” infrastructure26 |
Since 2016, economical assessments of various chemicals (e.g., carbon monoxide, ethylene, and alcohol products) have been made based on the development of catalysts.24–26 Note that the techno-economic analyses in Table 2 employ different evaluation metrics depending on the types of products. The solar to chemical products such as hydrogen and methanol are mainly evaluated through the ratio of produced energy to input energy, such as energy incorporation efficiency,26 energy return on energy invested,20 and energy payback time.20 On the other hand, the solar to chemical products are calculated using a cost-based metric such as net present value (NPV), levelized cost, and gross margin. Not surprisingly, these studies reported negative economic potential for solar-chemical production. However, the improvement on overpotential, one path conversion, and process efficiency should lower the cost of deploying solar-chemical systems. For example, Herron et al. argued that 45% CO2 one path conversion energy efficiency can be achieved.21
Herein, we report a state-of-the-art monolithic CO production PV–EC device and also present an NPV-based techno-economic analysis for a scale-up system. CO is one of the valuable products extracted from CO2 reduction, and it can be used in various industrial processes such as the Fischer–Tropsch process,29 alcohol synthesis,30 and ethylene glycol synthesis.31 CO is also an economically attractive product among various products extracted from CO2 reduction because the energy-price gap of CO (i.e., the price difference between the invested energy cost for production and the sales price) is also one of the highest.32 For the PV–EC device design, we used nanoporous silver and iridium oxide as electrocatalysts. Single-crystal silicon solar cells connected in series were designed for the PV photoelectrodes to provide sufficient power. The efficiency of the device was optimized by adjusting the PV photoelectrode to EC reactor area ratio and the electrolyte resistance. A commercial scale process design for photoelectrochemical CO2 reduction and a comprehensive economic analysis are presented in the following section. An electrolyzer model is built based on the monolithic device experimental result and gas conditioning processes such as the CO2 separation process are also proposed. The current status of the proposed PV–EC based CO production process is highlighted and sensitivity analysis shows the effect of important design variables.
Experimental
Preparation of PV modules
Photovoltaic modules were prepared using a commercially available interdigitated back contact (IBC) solar cell (SunPower Corporation, San Jose, CA). The cells were cut using a laser into 38 mm × 8.5 mm, 38 mm × 7.2 mm, and 38 mm × 6 mm sizes to form three types of mini-modules and were then connected in series using a ribbon cable. The prepared mini-modules were laminated between two layers of an encapsulating polymer, ethylene vinyl acetate, with a polycarbonate plate on both sides.Preparation of the nanoporous Ag catalytic electrode and IrOx oxygen evolution catalytic electrode
The nanoporous silver foil structure was prepared through electrochemical oxidation and reduction steps. First, the 5 × 5 cm2 silver foil (99.99%, LS Nikko) was oxidized to silver chloride at 0.3 V vs. an Ag/AgCl reference electrode for 12 h in 0.1 M KCl (99.0–100.5%, Sigma-Aldrich) electrolyte. Then the silver chloride layer on silver foil was reduced by applied potential of −1.2 V vs. Ag/AgCl in CO2 saturated 0.1 M KHCO3 (99.7%, Sigma-Aldrich) electrolyte solution. The IrOx film was prepared on Ti foil (Nilaco Co.) by the drop-casting method. The Ti foil was etched with 5 wt% oxalic acid (99%, Sigma-Aldrich) solution at 90 °C before IrOx coating. A 0.2 M H2IrCl6 (99.5%, Alfa Aesar) isopropanol solution was drop-cast on the Ti foil substrate, and then dried at 70 °C for 10 min. The foil was annealed at 500 °C in air, and the total process was repeated three times. Scanning electron microscopy (SEM, Hitachi-S4200) and X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe, Ulvac-PHI) were used to characterize the chemical state and morphology of the nanoporous Ag cathode and the IrOx coated Ti anode.Half-cell characterization of CO2 reduction and oxygen evolution for the nanoporous Ag catalytic film and IrOx catalytic film
Electrochemical characterization was carried out using a potentiostat (VSP, biologic) in a conventional three-electrode system with an Ag/AgCl reference and a platinum counter electrode. To characterize the CO2 reduction activity of the nanoporous Ag cathode, chronoamperometry was carried out in a closed cell with a constant applied potential, and a 0.1 M KHCO3 aqueous solution saturated with a continuous flow of CO2 gas was used as an electrolyte. The gaseous products were directly injected into the on-line connected gas chromatograph (GC) for the quantitative analysis, and the faradaic efficiencies were calculated from the measured current density and the volumetric concentration of each gaseous product was characterized by GC. Meanwhile, to characterize the oxygen evolution activity of the IrOx anode, linear sweep voltammetry was carried out with a 10 mV sec−1 scan rate in 1.0 M KOH.EC reactor assembly and electrochemical characterization
The EC reactor consisted of the cathode (nanoporous silver plate), the anode (IrOx coated Ti plate), and the bipolar membrane (fumasep FBM, Fumatech). Each component was assembled with a polycarbonate frame, and a silicon solar cell module was integrated into the EC reactor with a series connection. The catholyte (0.1–1.0 M KHCO3) was circulated between the cathode compartment of the EC reactor and an additional electrolyte reservoir while purging with CO2 gas. Similarly, the anolyte (1.0 M KOH) was separately circulated between the anode compartment and the anolyte reservoir while purging with Ar gas. To characterize the bulk electrolysis of the EC reactor, linear sweep voltammetry was carried out with a potentiostat (VSP, biologic) in a two-electrode configuration. Electrochemical impedance spectroscopy was carried out to measure the solution resistances based on a 10000 Hz to 1 Hz sigmoidal frequency at 3.5 V vs. cathode.
Measurement of the faradaic efficiency of CO and H2
During the electrochemical reaction, the gaseous products from a closed electrochemical half-cell or PV–EC device were directly injected into a gas chromatograph (YL6500, younglin) equipped with a Molsieve 13X packed column and Heysep D column. The detectors were a thermal conductivity detector and a flame ionization detector after a methanizer. The faradaic efficiency was calculated using eqn (1).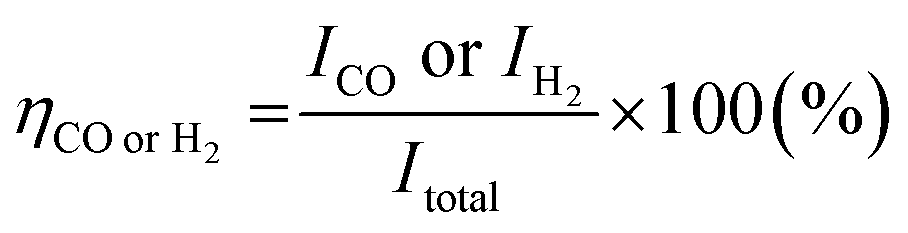
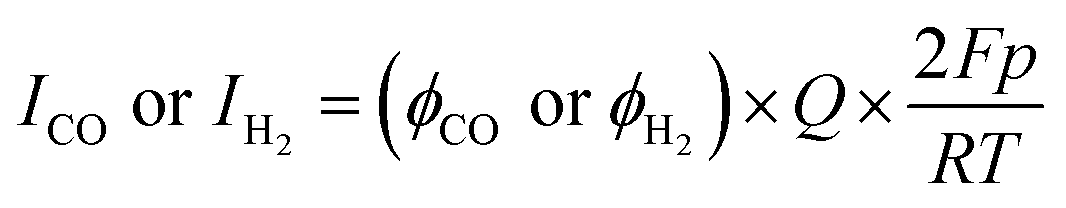 | (1) |
Results and discussion
Components of the PV–EC device and its design
Fig. 1 shows a general schematic of the proposed PV–EC unit cells and a photographic image of their parallel connected module. A PV–EC unit cell consists of components such as the catalyst, membrane, and solar cell. This configuration of the monolithic standalone PV–EC device has been demonstrated to have a high solar to CO conversion efficiency, ∼4.23% (ref. 33) by integrating the light absorbing semiconductor materials outside of the PV–EC cell. Nanoporous silver and iridium oxide were chosen as the CO2 reduction catalyst and water oxidation catalyst, respectively. Although the Au foil based catalyst has shown excellent faradaic efficiency and low overpotential,34 the high material cost makes the Au foil an inappropriate choice for industrial scale applications. Alternatively, a relatively lower priced material, silver, was chosen as the CO2 reduction catalyst. | ||
| Fig. 1 Solar to CO conversion device: image of the PV–EC module (left) and schematic of the PV–EC unit cell structure (right). | ||
Nanostructured silver-based catalysts also have an excellent faradaic efficiency (∼95%) for CO production.35–38 Earth-abundant materials such as Zn or modified carbon have also been suggested as efficient CO2 reduction catalysts; however, many of them exhibit a narrow potential window having high CO production selectivity from CO2 reduction compared to Ag based catalysts.37,38 In practical applications, the highest faradaic efficiency is desirable to get efficient CO/H2 separation process, and the narrow potential window causes difficulty to operation current matching an electrolyzer to a PV photoelectrode. The water oxidation (oxygen evolution) reaction, the counterpart of the CO2 reduction reaction (CO2RR), also requires a large overpotential. Iridium-based electrocatalysts are well known to have superior oxygen evolution activity, but they are also not cost-effective. Thus, iridium oxide nanoparticle layers were prepared on a titanium metal substrate to minimize the amount of iridium while retaining high catalytic activity. Ultimately, they should be replaced with cost-effective, earth-abundant material catalysts.
The PV photoelectrode was prepared using a silicon solar cell module to provide enough power for the EC reactor, and the Jsc and Voc of the silicon solar cell modules were controlled by the number of series connections made by the unit solar cells with the same active area (Fig. 2). These modules were labeled as Si-4, Si-5, and Si-6 according to the number of series-connected cells in a module. To determine a suitable specification for the PV photoelectrode, the catalytic activity of the cathode and anode must be considered. In our case, overpotentials of at least 600 mV and 296 mV were required for water oxidation and CO2RR, respectively to achieve ∼6 mA cm−2 resulting in a Voc of at least 2.3 V (i.e., 1.34 V theoretical electrochemical potential for the overall reaction + 0.9 V overpotential) excluding the effect of the solution resistance.
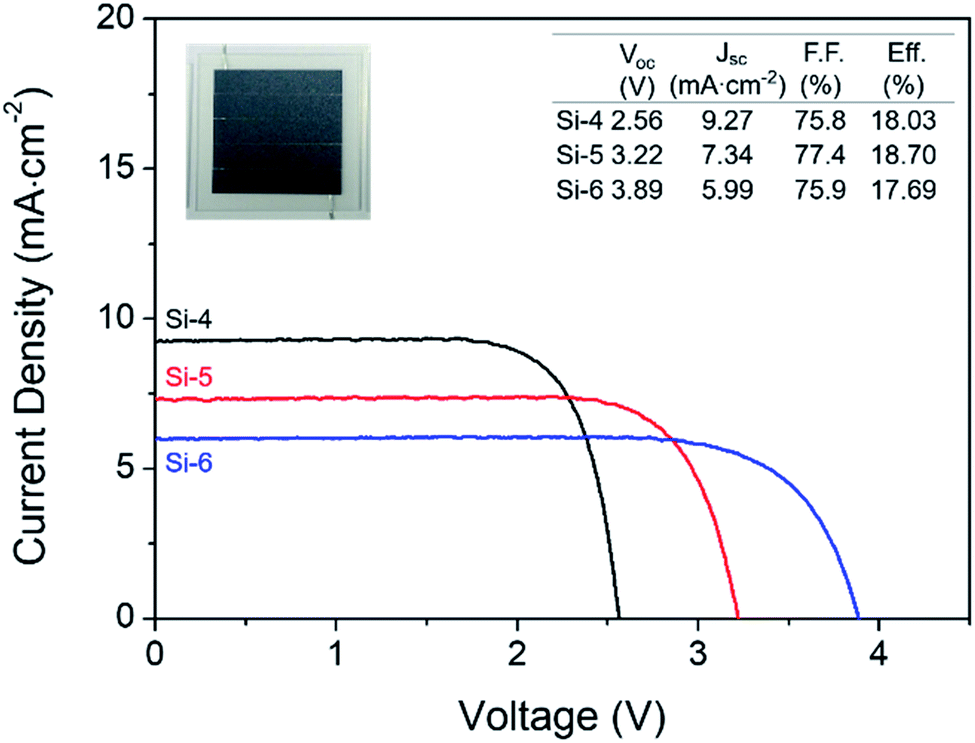 | ||
| Fig. 2 Individual I–V characteristic curves of photovoltaic modules for different numbers of unit cells. The inset table lists the open circuit voltage, short circuit current density, fill factor, and power conversion efficiency. The inset photograph is the Si-5 photovoltaic cell. | ||
Energy and conversion efficiency evaluation with device optimization
The equivalent series resistance in the EC cell can be affected by the conductivity of the electrolyte, or the distance between the electrodes. The voltage drop (IR drop) induced by the current flow (I) and resistance of the solution (R) is defined as Vdrop = I × R. Although this voltage drop reduces the performance of the EC reactor, it has not been considered as an important factor in solar-chemical production studies until the solar-to-chemical production current is large enough. However, when considering practical scales of PV–EC devices, solution resistance will not be a negligible factor. Many of the previous CO2 reduction electrocatalysts have been developed in 0.1–1.0 M bicarbonate electrolyte in order to yield high CO2 concentrations and to suppress the competitive hydrogen production reaction which produces high selectivity for the CO2 reduction reaction. However, these buffered electrolytes have low ionic conductivity, causing more significant loss due to the solution resistance.The solution resistance (R) is determined by several factors and can be expressed by eqn (2), where d is the distance between the electrode and separator (membrane), A is the electrode area, and κ is the conductivity of the solution.
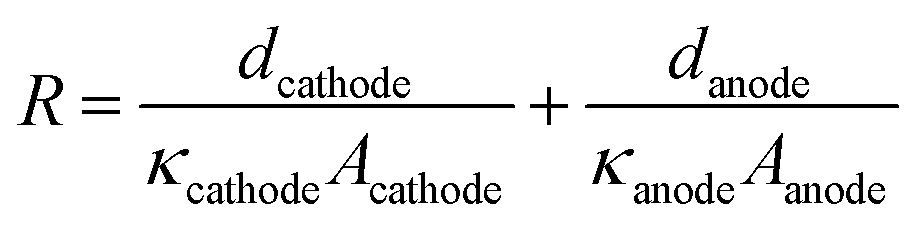 | (2) |
First, the gap distance of the cathode (Ag foil) from the membrane was controlled to investigate the effect of the gap distance on the PV–EC performance. The current density was increased with decreasing gap distance (Fig. 3(a)). When the current was compared at 3.5 V vs. cathode, the operating current was increased by almost two times (i.e. 45.9 to 81.6 mA) as the gap distance decreased from 22 to 8 mm, showing the importance of the EC reactor design. Minimizing the gap distance is required until the circulation of the electrolyte flow is allowed. The solution resistance of the two-electrode system was measured at each gap distance by electrochemical impedance spectroscopy based on the equivalent circuit depicted in (Fig. S3(b),† linear sweep voltammetry (LSV) results are also available in Fig. S1†).
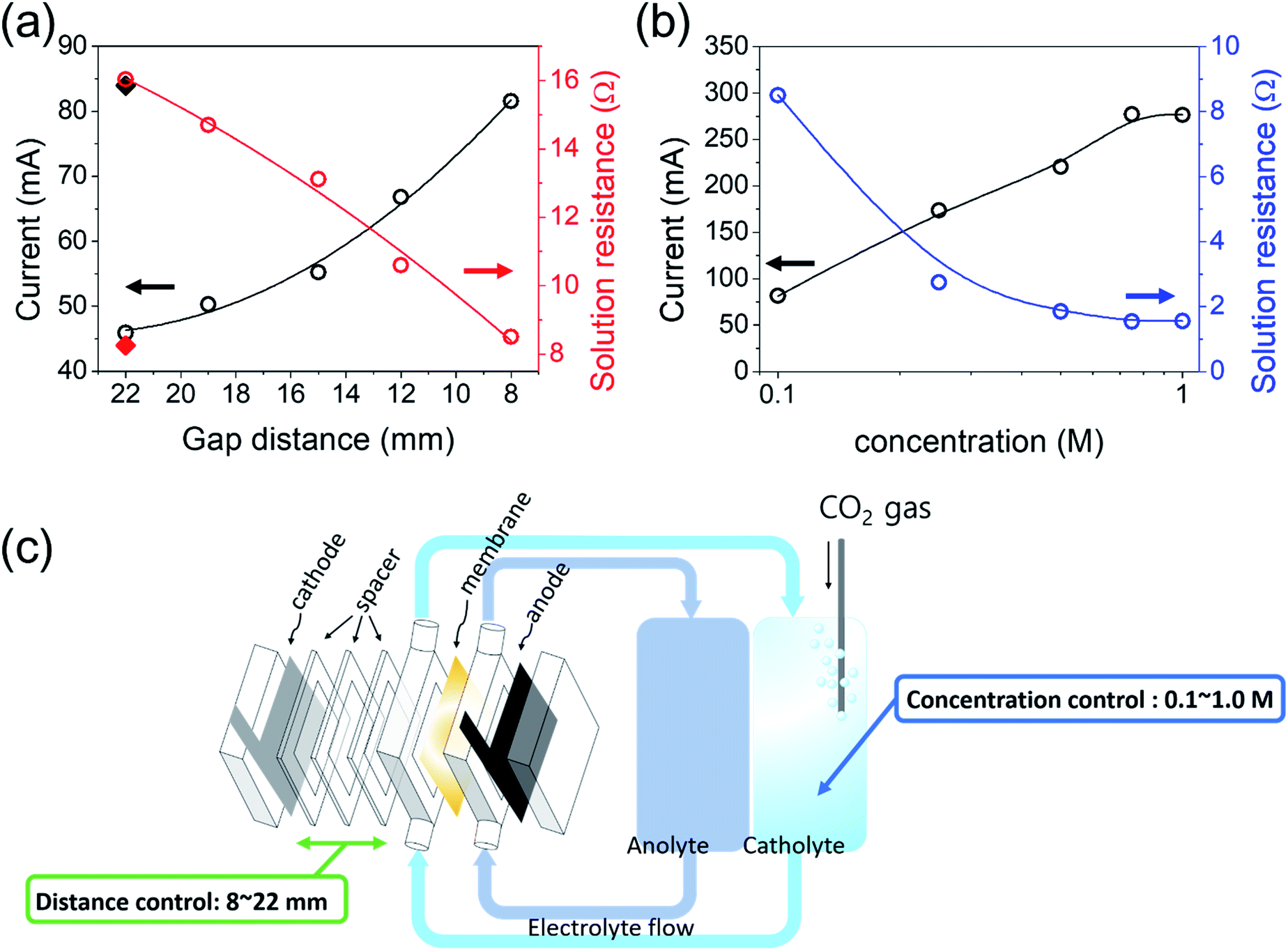 | ||
| Fig. 3 The current and solution resistance behaviours with respect to the (a) electrode–membrane gap distance and (b) electrolyte concentration. (c) Schematic description of EC reactor performance investigation through the control of the electrode–membrane gap distance and electrolyte concentration. | ||
Fig. 3(b) shows the current and the solution resistance relationship with respect to the concentration variation. In our system, the conductivity of the 1 M KOH anolyte solution is high enough (178 mS cm−1) compared to the catholyte (0.1 M KHCO3, pH 6.78, 4.6 mS cm−1).39 Therefore, the total solution resistance is majorly affected by the cathode side. To reduce the solution resistance of the catholyte further, the concentration of KHCO3 was increased, because a higher ionic concentration increases the conductivity of the solution. Note that the bipolar membrane was used to minimize the membrane potential for a pH gradient system. The theoretical overall reaction potential of CO2 + 2H2O → CO + O2 + 2H2O is 1.34 V in the absence of a pH gradient, but it can be reduced when a pH gradient is created. However, at the same time, the membrane potential is increased with increasing pH gradient; therefore, there is no overall potential variation due to the pH gradient.40–42 Similar to the gap distance control effects, the current proportionally increased with the KHCO3 concentration at 3.5 V vs. cathode (the solution resistance and linear sweep voltammetry (LSV) results are available in Fig. S4†). However, the current was saturated at 0.75 M of CO2-saturated KHCO3, showing a very similar LSV result between 0.75 and 1.0 M of KHCO3. This result may be attributed to various factors including the pH of the solution and the solubility of CO2. The current is created by both H2 and CO generation; thus, it depends on both pH and CO2 concentration. KHCO3 concentrations higher than 1.0 M in electrolyte may not be appropriate to efficiently perform the CO2 reduction reaction owing to the low CO2 concentration in a high pH solution. It is worth noting again that this solution resistance may not be critical when the solar-to-chemical production current was small; however, it will become significant as the current or conversion efficiency increases.
Fig. 4(a) shows the summarized results of the proposed device sensitivity depending on the electrode distance or electrolyte concentration in addition to the type of Ag catalyst (e.g. flat or nanoporous Ag foil). The nanoporous Ag showed a reduced overpotential of approximately 350 mV (Fig. S3(a) and (b)†) (the detailed characterization data of nanoporous Ag and IrOx electrodes are also included in Fig. S3 and S4†); therefore, the LSV of the EC reactor was also cathodically shifted by approximately 350 mV. Fig. 4(a) also shows an estimation of the optimum series connections between the PV.
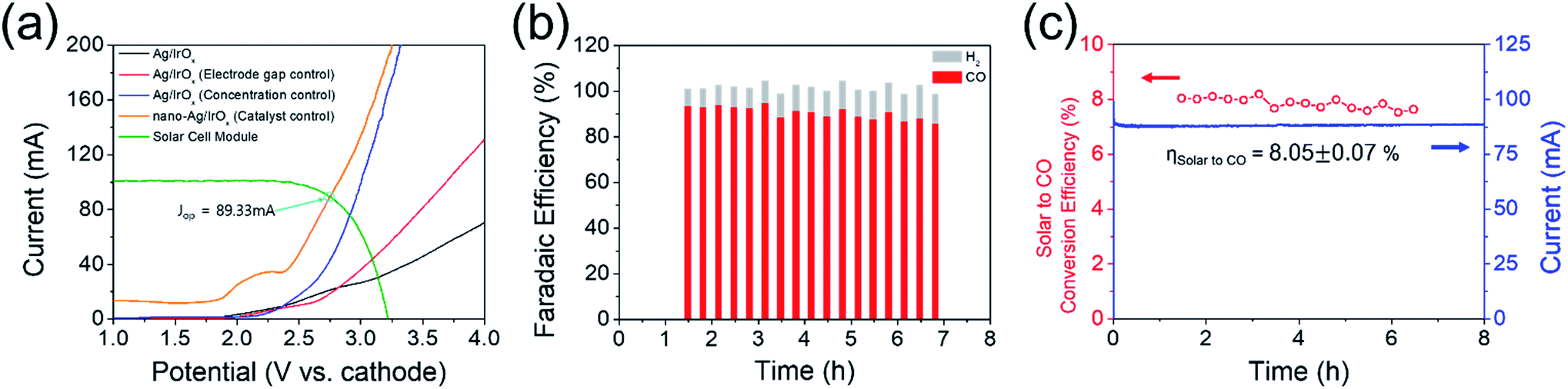 | ||
| Fig. 4 (a) Optimization of operating I–V parameters of the EC reactor with respect to various factors when the Si-5 solar cell was used as a PV photoelectrode. (b) Faradaic efficiency behaviour with respect to the PV–EC device operation time. (c) Solar to chemical conversion efficiency and steady state current during PV–EC device operation. | ||
As expected from our PV photoelectrode design, three PV photoelectrodes (Si-4, 5, and 6) were suitable for the EC reactor with nanoporous Ag/IrOx catalysts (Fig. 4(a)). However, the highest Iop was obtained from the PV–EC device with the Si-5 PV photoelectrode. Using this device, the steady state current obtained under the standard illumination condition (1 Sun, 1.5 AM) was very similar to the expected Iop value (Fig. 4(b) and (c)). The faradaic efficiency of the product was measured during the reaction. The major gaseous product was CO (93%), and the remainder was H2 (7%), which is also similar to the result obtained from the closed half-cell experiment using the nanoporous Ag catalyst. The solar to CO conversion efficiency was estimated during the reaction time (Fig. 4(c)) based on eqn (3).
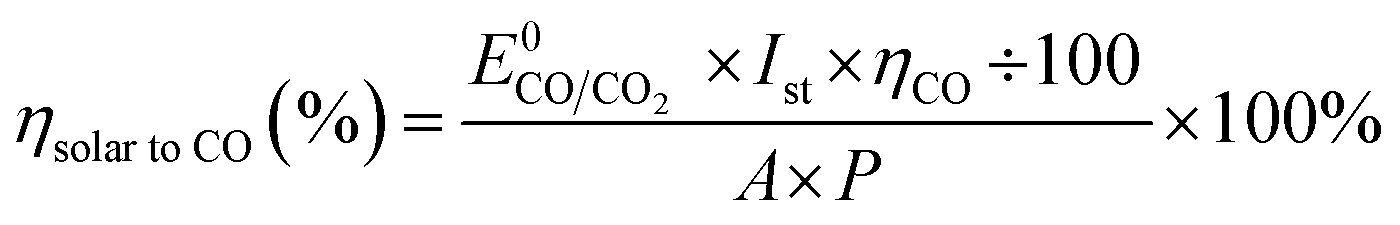 | (3) |
| a Solar to CO conversion efficiency. b Calculated from given values in the reference. c Solar to formate conversion efficiency. d Formate production rate photoelectrode and the EC reactor. The intersection of the I–V curve (Iop) indicates the operating current of our PV–EC device. | ||||
|---|---|---|---|---|
| This work (KIST) | Schreier et al. (EPFL)12 | Schreier et al. (EPFL)10 | Zhou et al. (Caltech)8 | |
| Solar to chemical efficiency (%) | 8.05a | 6.5a | 13.4a | 10c |
| PV efficiency (%) | 18.7 | 13.4 | 28.5 | — |
| EC reaction efficiency (%) | 44.61 | 48.5 | 47 | — |
| Production rate (mL h−1)b | 37.7 | 0.57 | 2.72 | 1.70 × 10−5d |
| Photovoltaic cell | c-Si solar cell | Perovskite solar cell | GaInP/GaInAs/Ge 3-junction solar cell | GaAs/InGaP/TiO2/Ni photoanode |
| Cathode material | Nanoporous Ag | OD – Au | SnO2/CuO | Pd/C/Ti |
| Anode material | IrOx | IrO2 | SnO2/CuO | GaAs/InGaP/TiO2/Ni |
| Photon absorber/electrode area | Photovoltaic: 13.68 cm2 | Photovoltaic: 0.2858 cm2 | Photovoltaic: 0.2858 cm2 | Photoanode: 0.03 cm2 |
| Cathode: 19.27 cm2 | Cathode: 1 cm2 | Cathode: 1 cm2 | Cathode: 0.04 cm2 | |
| Anode: 19.27 cm2 | Anode: 4.5 cm2 | Anode: 4.5 cm2 | ||
Although some devices showing higher than 10% efficiency have been achieved, they generally require highly expensive III–V solar cell technology. The solar to CO conversion efficiency can be evaluated by the product of the EC reaction efficiency and PV photoelectrode efficiency, so a highly efficient PV application is desirable. On the other hand, there is room for enhancing the solar to CO conversion efficiency by increasing the EC reaction efficiency. The EC reaction efficiency can be enhanced by applying higher-efficiency catalytic electrodes. Furthermore, the internal resistance (especially solution resistance) in a device significantly affects the overall EC reaction efficiency, which is strongly dependent on the electrode size (reactor size). Considering the solar to CO conversion efficiency obtained by small electrodes in most previous studies, our EC reaction efficiency has achieved a very promising value, implying huge potential for practical applications. The production rate of CO was also calculated using eqn (4).
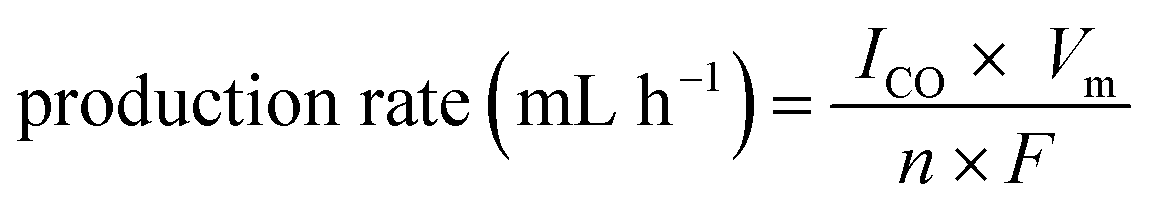 | (4) |
The conversion of the PV–EC device was also investigated by changing the flow rate of the CO2 saturated electrolyte. In general, the CO2 electro-reduction activity of an electrocatalyst or device has been tested in a batch reactor with a CO2 saturated electrolyte or continuous CO2 gas flow condition. Continuous flow reactors are favoured in industrial processes owing to their large production capacity. In this type of reactor, the CO2 concentration is kept constant during the reaction, and the production rate can be controlled by changing the feed rate of the reactant. In our system, the electrolyte in the reservoir is saturated by CO2 purging and is circulated using a water pump. In this case, the CO2 gas was not directly supplied to the EC reactor; only dissolved CO2 was provided to the reactor. Despite different flow rates (1–100 mL min−1) the total current during light irradiation did not vary significantly (Fig. 5(b)). In contrast, the selectivity (faradaic efficiency) of CO and H2 was strongly dependent on the flow rate, which is consistent with the results from the previous study where the layer thickness variation of the mass transfer boundary influenced by the electrolyte flow rate was suggested as a main reason.43,44
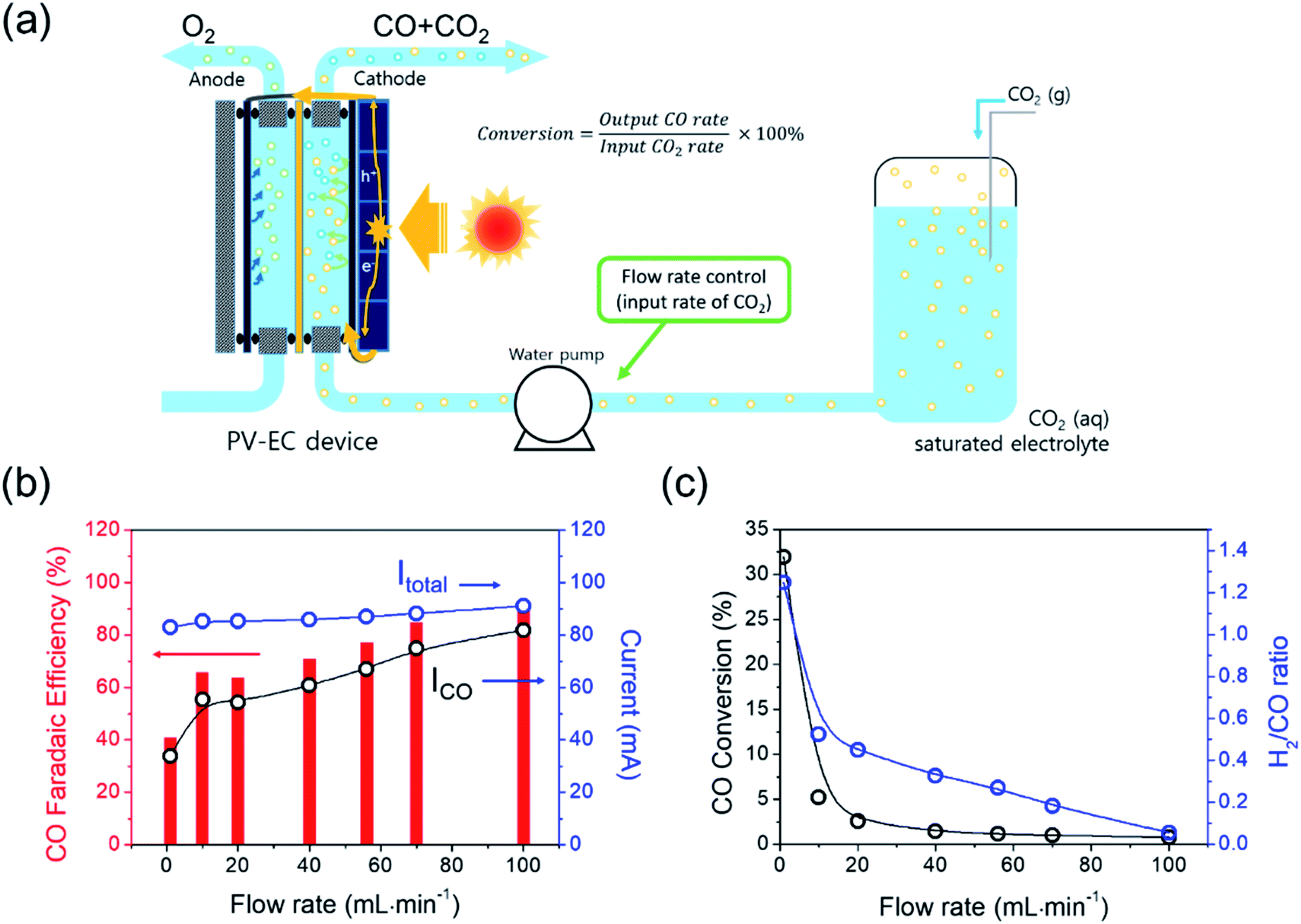 | ||
| Fig. 5 (a) Schematic description of conversion estimation for the PV–EC system. (b) Total current and partial CO current, and CO faradaic efficiency with respect to the electrolyte flow rate. (c) CO2/CO conversion and H2/CO ratio results with respect to the flow rate. | ||
The electrochemical CO2RR is directly related to the mass transfer boundary layer thickness at the cathode surface (an electrochemical double layer), rather than to the bulk concentration of CO2. The CO2 (aq) is mainly depleted in the near surface of the electrode while the CO2 (aq) concentration of the bulk electrolyte (0.033 M) essentially remains constant.13,45 The remainder of the partial current was used for H2 generation, and the total faradaic efficiency was approximately 100% at any flow rate. The CO conversion was calculated from the experimental data and using eqn (5).
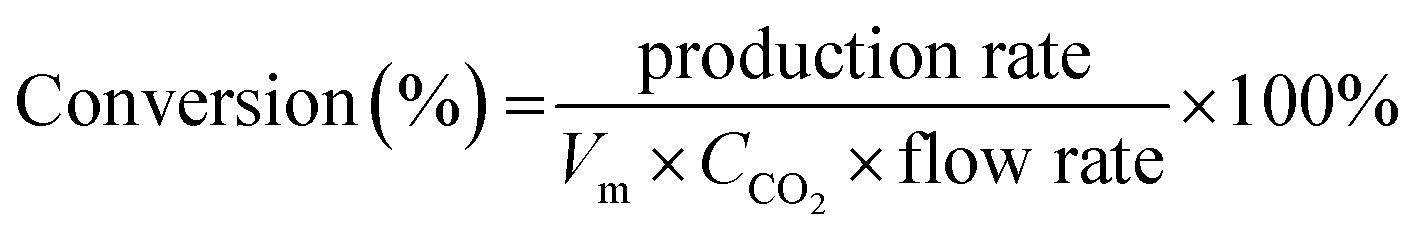 | (5) |
Process design and techno-economic analysis
The conceptual process design and techno-economic analysis of a scaled-up CO production process were carried out to evaluate the economic feasibility of the proposed PV–EC device. Fig. 6 shows the process flow diagram of the CO production process. The CO production process consists of a CO2 saturation column (D110), a CO separator (D130), an O2 separator (D120), two pumps (P140 and P150), and an EC reactor (R160). The CO2 saturated solvent (stream (4)) is pumped into the EC reactor (stream (5)), where the EC reaction takes place. The phase separation of the CO product stream (6) is carried out in a CO flash separator (D130). Similarly, the water stream (11) is pumped to the electrolyzer (R160) where O2 is produced through water oxidation. Because the anode product stream (9) mainly consists of the anolyte and O2, a simple flash separator (D120) can recover O2 (13). The fraction of CO in the vapor product stream (8) varies depending on the conversion of the CO2 in the EC reactor, and the vapor stream also includes excess amounts of CO2 when the conversion rate is low. Note that the proposed CO production system is operated under atmospheric pressure and its CO2 conversion rate is approximately 1.4%.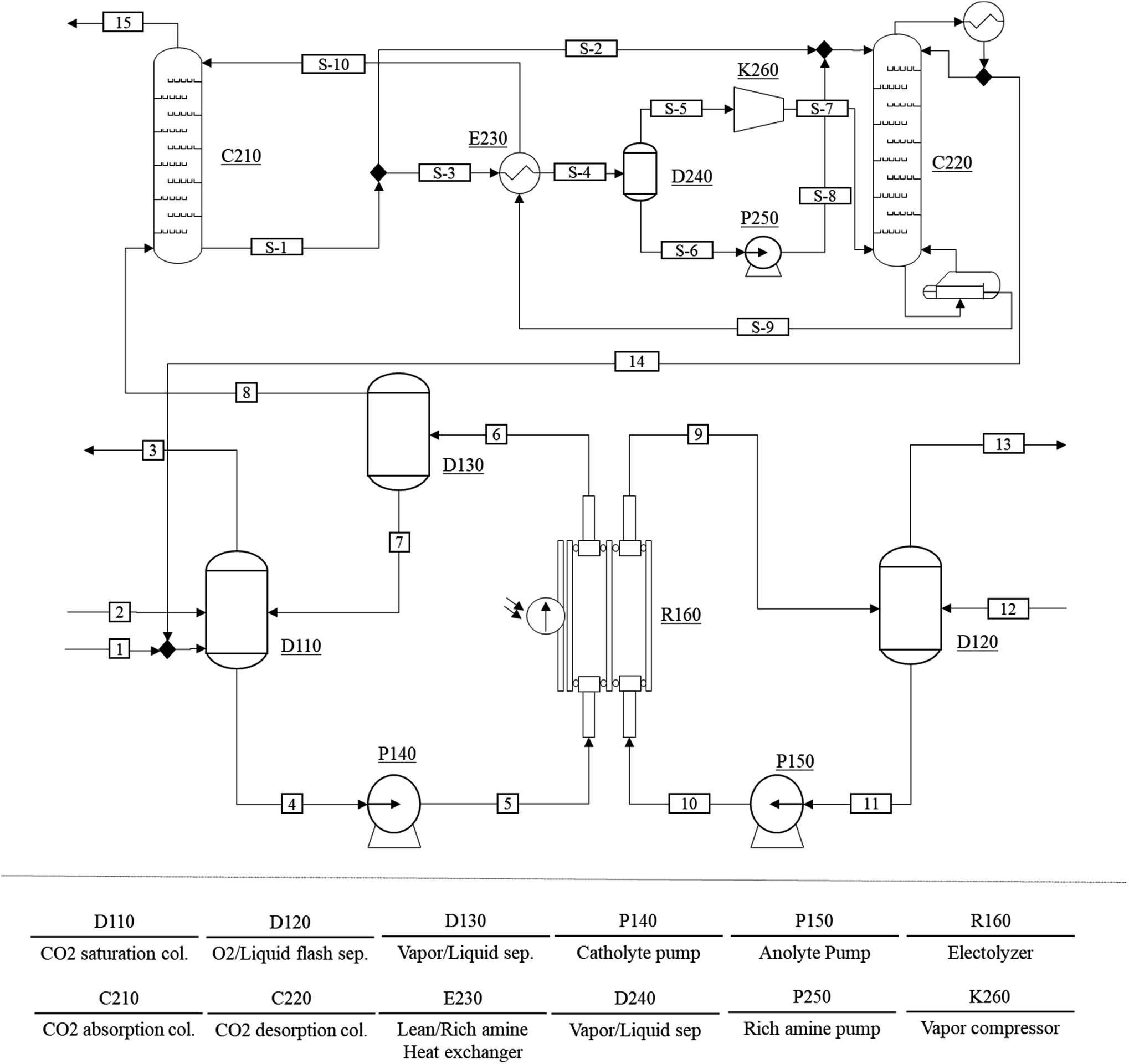 | ||
| Fig. 6 Process flow diagram of the electrochemical CO production system. | ||
Consequently, the vapor product stream includes 79 mol% of CO2. An additional gas separation process (C210 ∼ K260) is employed to remove the CO2 from the product. The CO2 separation process initially proposed by Lee et al. is modified for the purification of the CO product.19 In the CO2 separation process, an aqueous amine solvent is used to selectively remove CO2 from the vapor product stream (8). Because the amine solvent chemically reacts with CO2 (forming carbamate and bicarbonate), the CO2 having low partial pressure can be effectively removed from the product stream. The rich amine vapor recompression with the split input flow configuration proposed by Jung et al. is also included to minimize the energy consumption of the gas separation step.46 The recovered CO2 is recycled back to the CO2 saturation column to minimize the raw material supply of the process.
| CO2 + 2H+ + 2e− → CO + H2O | (6) |
| 2H+ + 2e− → H2 | (7) |
| 2H2O → O2 + 4H+ + 4e− | (8) |
| CO2 → CO + 1/2O2, ΔHrxt = 283 kJ mol−1 | (9) |
| H2O → H2 + 1/2O2, ΔHrxt = 242 kJ mol−1 | (10) |
| ṁ5 × CCO2 × X × ΔHrxt,CO2 = V × Jop,CO2 × Aelec | (11) |
The Peng–Robinson equation of state is used to precisely predict the phase equilibrium and enthalpy of the process streams. Binary interaction parameters between water and CO2 are regressed by using the maximum likelihood equation. Vapor liquid equilibrium experimental data were obtained from Fonseca et al.47 and Schüler et al.48 Detailed results and verification of the regression are available in the ESI, Table S1, and Fig. S5.†Eqn (6) and (7) describe the electrochemical reduction of the cathode side and eqn (8) specifies the water oxidation on the anode side of the EC reactor. To simplify the EC reactor model, these EC reactions are reduced; thus, reaction enthalpies are calculated as state functions. ṁ, C, X, and Aelec respectively denote the mass flow rate, concentration, conversion, and electrode area. We assume that the partial CO2 current density (Jop; CO2) and operating EC reactor voltage (Voc) are the same as in the lab-scale experiment. The conversion of CO2 can be calculated using eqn (11). We compared the CO2 conversion for the experiment and the EC reactor simulation under the experimental conditions to validate the proposed model. The experiment achieved 1.45% CO2 conversion at atmospheric pressure, 35 °C, and 33 mM CO2 concentration, while the process simulation predicted 1.37% CO2 conversion. The difference results from the ideal reaction assumption; however, the proposed model shows good agreement with the experimental results.
Table 4 lists the financial parameters used to evaluate the cost of the proposed CO production process. We assume 335 days of process operation per year and 5.57 h per day operation to satisfy the solar capacity factor. A 1.2 m2 PV cell having 225 W capacity is adopted in this study. The equipment purchase cost of the electrochemical CO production system is based on the 2018 U.S. market prices. Conventional unit operations such as columns, pumps, and heat exchangers are estimated using the Guthrie correlation and are updated using the 2018 chemical plant cost index.49 Detailed explanations and MATLAB codes for sizing and costing the equipment adopted in this study are available in Lee et al.19 The unit price for nonsubsidized single crystalline Si PV cells is taken from the 2018 market prices. The unit price of the EC reactor is calculated based on our experiment and is estimated at $7200 per m2. Among the constituents of the EC reactor, the bipolar membrane is the most expensive component and represents 44% of the total unit cost.
| Parameter | Value |
|---|---|
| Plant life time (years) | 20 |
| Solar capacity factor | 0.232 |
| Interest rate | 10 |
| $ basis year | 2018 |
Table 5 summarizes the equipment purchase costs of a 4 MW based PV–EC CO production process. The total PV cell cost including the panel mounting and hard balance of the system is approximately $2.7 million and corresponds to $192 per 1.2 m2 panel. Note that the PV cells contribute less than 2% of the total equipment cost. The recent price decrease in single crystalline Si solar cell panels makes them economically applicable for the artificial photosynthesis process. The EC reactor, however, is far more expensive for the commercial application of the process. In fact, the EC reactor is responsible for more than 90% of the total equipment cost because of the expensive bipolar membrane, Ag, and IrOx catalyst. The equipment costs of the PV cell and the EC reactor are linearly dependent on the CO production scale. On the other hand, the scale-up process is advantageous for conventional unit operations involving saturation columns, pumps, and the CO2 separation process. Note that the contributions of the PV system, auxiliary unit operations, and CO2 separation system can be as high as 70% when the CO2 production system is scaled down to a 1.2 m2 single PV cell system.
| Component | Equipment cost (M$) |
|---|---|
| Photovoltaic module | 1.5 |
| Panel mounting | 0.4 |
| Other PV BOS | 0.8 |
| PV total | 2.7 |
| Electrolyzer | 219.0 |
| Wiring | 0.2 |
| Other electrolyzer BOS | 0.9 |
| Electrolyzer total | 220.1 |
| Cathode saturation column | 0.92 |
| Anode saturation column | 0.92 |
| CO separator | 0.92 |
| Cathode pump | 0.02 |
| Anode pump | 0.02 |
| Reaction system process unit total | 2.79 |
| CO2 separation system | 2.4 |
| Total | 228.0 |
The total annualized cost (TAC) is calculated to obtain the minimum sales price of CO. The TAC is composed of the annualized investment cost (AIC) and the average annualized operating cost (AOC). The AIC comprises the direct cost, indirect cost, working capital, and the start-up cost. The AIC is annualized over 20 years with 10% interest rate and is added to the AOC to determine the TAC. A pre-tax environment is assumed; thus, depreciation is not considered for the capital assets. Here, we include the raw material cost and the CO2 purification cost in the AOC to determine the simplified TAC; however, operating labor cost, plant overhead, and general expenses should also be considered for more detailed analysis in the future. In addition, the 20 years of plant operation is an optimistic assumption because the electrolysis system generally shows far less durability. Note that we assume $40 per ton CO2 raw material cost and negligible water cost.19 The equivalent cost of CO is calculated by the annualized worth (AW) method50 adjusting its sales prices such that the present worth reaches zero (eqn (12)) where DCO and PCO respectively indicate the demand and price of CO.
| AW = 0 = DCO × PCO − AOC − AIC | (12) |
The energy and mass balance table of the solar driven CO production process is available in Table S2.† The 4 MW PV–EC system corresponds to 3.4 ton per day CO production. The proposed process generates 22.3 MWhe per day for CO production and it is converted to either the 3.0 × 104 m2 PV module area or to 1.8 × 104 for the 1.2 m2 unit PV modules. The amount of solvent stream flowing into the EC reactor is considerably higher than the CO2 stream. The high flow rate results from the low CO2 concentration and conversion, yielding a large recycling stream of CO2. The vapor product stream (stream 8) contains 79 mol% CO2; hence, an additional gas separation process is unavoidable for the recovery of a high purity CO stream. However, the high fraction of CO2 in the product stream can be lowered as the CO2 conversion increases, consequently lowering CO purification costs. Thus, a method for feeding a more concentrated CO2 stream and/or achieving higher conversion should be developed in future studies to lower the capital cost of the process.
Table 6 summarizes the total capital expenditure. The cost of each item is evaluated using the parameters proposed by Douglas.51 Given the total capital expenditure, the AIC and yearly CO production are estimated at approximately $11.6 M and 1.1 kton per year, respectively. The high AIC mainly results from the expensive equipment. Thus, technological improvements or replacement of expensive elements in the EC reactor may reduce the purchase equipment cost significantly and save some of the capital expenditure for improved commercialization. The raw material (i.e., water and CO2) costs are only $0.47 M because most of the CO2 feed is supplied by the large recycle stream from the gas separation process. The annual operating cost for gas separation is $0.37 M in the amine-based CO2 capture process. The TAC is calculated as $12.5 M and corresponds to the CO sales price of $10.94 per kg. Note that the net amount of CO2 reduction is approximately 1.8 kton per year. The solar driven CO production process is intrinsically limited by low solar power density, solar capacity factor, and conversion. These limitations, together with the high total capital expenditure, result in a sales price for CO that is approximately 18 times the price for CO produced in commercial fossil fuel driven processes ($0.6 per kg CO25). Accordingly, the solar driven CO production process proposed in this study should not be expected to compete with the fossil fuel driven CO market unless the CO2 tax reaches approximately $6.6 per kg. Given the equipment purchase cost of the EC reactor, we analyze the cost contribution of each element (i.e., Ti support, Ag catalyst, IrOx catalyst, membrane, and frame).
| Fraction FCI | Used | Cost (M$) | |
|---|---|---|---|
| Direct cost | |||
| ISBL | 60.0% | 345.97 | |
| Purchased equipment | 20–40% | 40.0% | 230.65 |
| Purchased equipment installation | 7.3–26% | 12.0% | 69.19 |
| Instrumentation and control | 2.5–7.0% | 2.5% | 14.42 |
| Piping | 3–15% | 3.0% | 17.30 |
| Electrical | 2.5–9.0% | 2.5% | 14.42 |
| OSBL | 20.0% | 115.32 | |
| Building and building services | 6–20% | 8.0% | 46.13 |
| Yard improvements | 1.5–5.0% | 2.0% | 11.53 |
| Services facilities | 8.0–35.0% | 8.0% | 46.13 |
| Land | 1–2% | 2.0% | 11.53 |
| Total direct cost | 80.0% | 461.29 | |
|
|||
| Indirect cost | |||
| Engineering | 4–21% | 6.0% | 34.60 |
| Construction expenses | 4.8–22.0% | 7.0% | 40.36 |
| Contractor's fee | 1.5–5.0% | 2.0% | 11.53 |
| Contingency | 5–20% | 5.0% | 28.83 |
| Total indirect cost | 20.0% | 115.32 | |
| Fixed capital investment (FCI) | 100 | 100.0% | 576.61 |
| Working capital | 10–20% | 10.0% | 57.66 |
| Start-up cost | 8–10% | 8.0% | 46.13 |
| Total capital investment (CAPEX) | 680.41 | ||
As shown in Fig. 7, the membrane is the most expensive element for EC reactor manufacturing followed by the IrOx catalyst, and although the PV–EC CO production process is far more expensive at the current technology level, it is still a promising technology, especially for CO2 utilization. The process actually consumes CO2 as a raw material with almost no CO2 generation during the process and can yield an order of magnitude higher sales margin compared to PV electricity generation. In addition, Fig. 7 indicates that a lower cost membrane and/or cathode catalyst can facilitate the commercialization of the CO production in the EC reactor. Higher solar to chemical efficiency, conversion, and higher current density can also improve the economic feasibility of a photoelectrochemical CO production system.
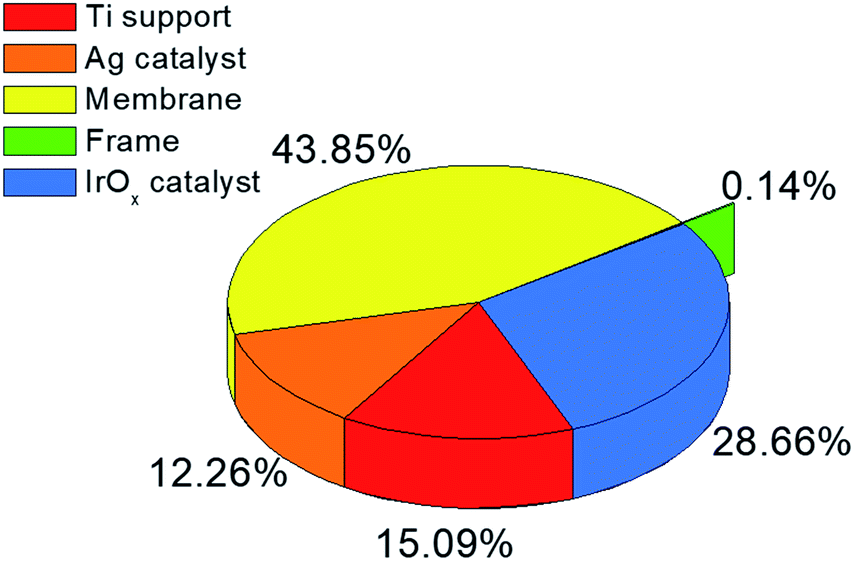 | ||
| Fig. 7 electrochemical (EC) reactor element costs. | ||
Fig. 8 shows the equivalent CO sales price changes depending on the EC reactor current density, the CO2 conversion, the solar to CO efficiency, and the relative membrane price. The relative membrane price, here, is defined as the price ratio to the current membrane purchase price (i.e., a relative membrane price of 0.8 indicates 80% of the current membrane price). This analysis shows that improving the current density and the conversion rate has the largest impact on the CO sales cost which can even reach the fossil fuel driven CO sales price. The white solid line indicates the equivalent prices that make the CO sales cost the same as the commercial CO sales cost. When the EC reactor current density is higher than 80 mA cm−2, economically profitable CO production can be achieved. However, approximately 22% CO2 conversion is required; thus, technological improvements that suppress the H2 side reaction at a low feed rate are necessary. When the current density is sufficiently high (>120 mA cm−2), economically feasible CO production is possible even with the CO2 conversion rate of 10%. An improvement in current density or conversion rate leads to decreasing size of the EC reactor, and thus reduces the capital expenditure for the solar driven CO production process. Fig. 8(b) shows the influence of the membrane cost and the solar to CO efficiency. The equivalent CO production cost is also significantly lowered as a less-expensive membrane and/or a higher efficiency device are introduced. For example, the equivalent CO production cost of $10.94 per kg can be lowered to $2.7 per kg when the solar to CO efficiency increases to 20%, and the membrane cost is reduced to half of the current price. Note that the equivalent CO production cost is still higher than the current CO sales price within the search area; thus, the CO2 conversion and current density improvement study should be conducted together with the membrane cost and efficiency improvement.
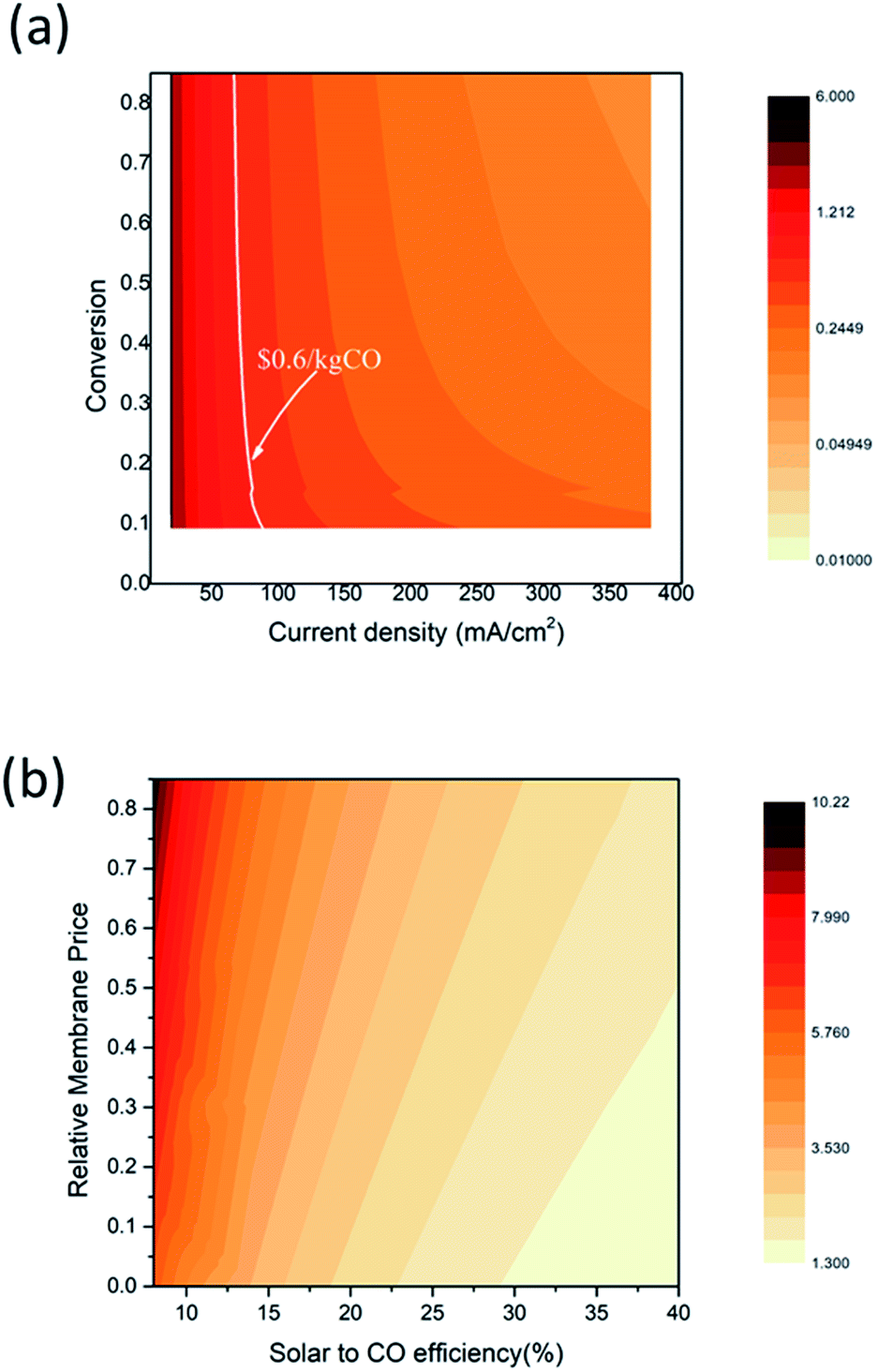 | ||
| Fig. 8 Contour plot of the equivalent CO sales cost. (a) CO sales cost depending on the EC reactor current density and CO2 conversion. The white solid line indicates the market price of CO ($0.6 per kg CO25) (b) CO sales cost depending on the solar to CO efficiency and the relative membrane price. | ||
Conclusions
This paper presents a state-of-the-art solar-chemical production device and comprehensive economic analysis for the commercial scale CO production process. The proposed PV–EC device shows 8.03% solar to CO conversion efficiency and 0.77 to 31.9% CO2 conversion with the device optimization. In addition, we also demonstrate the CO/H2 ratio control using the reactant flow rate, and thus the desirable chemicals (e.g., syngas or high purity CO) can be produced by changing process operating conditions. Techno-economic analysis shows that the equivalent CO production cost is about $10.94 per kg using the current technology. However, technological improvements on the membrane, device current density, and CO2 conversion can facilitate the practical application of the solar to chemical production technology in the near future. Typically, the proposed system may be economically feasible when high current density (>120 mA cm−2) is achieved along with more than 10% one path CO2 conversion under optimistic assumptions (e.g., Vcell < 2.75, FE > 93%, and durability > 20 years). Several research studies can be suggested for the future study. For example, electrocatalyst materials and membranes having low price should be developed to compete with commercialized CO production processes. In addition, the optimization of the PV/EC device area in terms of the cost is necessary because it is desirable to reduce the expensive EC reactor cost. Finally, the operation strategies of the process under the dynamic solar energy supply should be carefully examined.Author contribution
S. Y. C. performed most of the experiments including preparation of the catalyst electrode and electrochemical measurements. S. Y. L. constructed the gaseous product quantification system. S. G. H. contributed to the synthesis of the IrOx catalyst. O. S. J. and H. K. contributed to the interpretation of experimental data. J. K., S. P., D. K., and Y. K. fabricated the PV photoelectrode. U. L. carried out techno-economic analysis. B. K. M. and Y. J. H. designed and supervised the project. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Institute of Science and Technology (KIST) institutional program, by “Next Generation Carbon Upcycling Project” (Project No. 2017M1A2A2046713) through the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Republic of Korea, and by the KU-KIST Graduate School Project.Notes and references
- J. A. Turner, Science, 1999, 285, 687–689 CrossRef CAS PubMed.
- U. N. G. Assembly and W. C. o. Environment and Development, Our Common Future, WCED, 1987 Search PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- S. R. Lingampalli, M. M. Ayyub and C. N. R. Rao, ACS Omega, 2017, 2, 2740–2748 CrossRef CAS PubMed.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- U. Kang, S. K. Choi, D. J. Ham, S. M. Ji, W. Choi, D. S. Han, A. Abdel-Wahab and H. Park, Energy Environ. Sci., 2015, 8, 2638–2643 RSC.
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis and C. Xiang, ACS Energy Lett., 2016, 1, 764–770 CrossRef CAS.
- Gurudayal, J. Bullock, D. F. Srankó, C. M. Towle, Y. Lum, M. Hettick, M. C. Scott, A. Javey and J. Ager, Energy Environ. Sci., 2017, 10, 2222–2230 RSC.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS.
- J. T. Song, H. Ryoo, M. Cho, J. Kim, J.-G. Kim, S.-Y. Chung and J. Oh, Adv. Energy Mater., 2017, 7, 1601103 CrossRef.
- M. Schreier, L. Curvat, F. Giordano, L. Steier, A. Abate, S. M. Zakeeruddin, J. Luo, M. T. Mayer and M. Grätzel, Nat. Commun., 2015, 6, 7326 CrossRef CAS PubMed.
- M. R. Singh, J. D. Goodpaster, A. Z. Weber, M. Head-Gordon and A. T. Bell, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8812–E8821 CrossRef CAS PubMed.
- J. Brillet, J.-H. Yum, M. Cornuz, T. Hisatomi, R. Solarska, J. Augustynski, M. Graetzel and K. Sivula, Nat. Photonics, 2012, 6, 824 CrossRef CAS.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, J. Ma, Z. Zhang, G. Yang and B. Han, Green Chem., 2017, 19, 2086–2091 RSC.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- J. L. Young, M. A. Steiner, H. Döscher, R. M. France, J. A. Turner and T. G. Deutsch, Nat. Energy, 2017, 2, 17028 CrossRef CAS.
- M. Yamaguchi, Sol. Energy Mater. Sol. Cells, 2003, 75, 261–269 CrossRef CAS.
- U. Lee, J. Burre, A. Caspari, J. Kleinekorte, A. M. Schweidtmann and A. Mitsos, Ind. Eng. Chem. Res., 2016, 55, 12014–12026 CrossRef CAS.
- R. Sathre, C. D. Scown, W. R. Morrow, J. C. Stevens, I. D. Sharp, J. W. Ager, K. Walczak, F. A. Houle and J. B. Greenblatt, Energy Environ. Sci., 2014, 7, 3264–3278 RSC.
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126–157 RSC.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti, G. Harrison and E. Tzimas, Int. J. Hydrogen Energy, 2016, 41, 16444–16462 CrossRef.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- S. Verma, B. Kim, H.-R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- C. Palmer, F. Saadi and E. W. McFarland, ACS Sustainable Chem. Eng., 2018, 6, 7003–7009 CrossRef CAS.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- S. Hernández, M. A. Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326–2346 RSC.
- M. E. Dry, J. Chem. Technol. Biotechnol., 2002, 77, 43–50 CrossRef CAS.
- K. Fang, D. Li, M. Lin, M. Xiang, W. Wei and Y. Sun, Catal. Today, 2009, 147, 133–138 CrossRef CAS.
- K. Dong, S. Elangovan, R. Sang, A. Spannenberg, R. Jackstell, K. Junge, Y. Li and M. Beller, Nat. Commun., 2016, 7, 12075 CrossRef PubMed.
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, Nanotechnology 2011: Bio Sensors, Instruments, Medical, Environment and Energy, NSTI, 2011 Search PubMed.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- L. Q. Zhou, C. Ling, M. Jones and H. Jia, Chem. Commun., 2015, 51, 17704–17707 RSC.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, 96th Edition, CRC Press, 2015 Search PubMed.
- D. A. Vermaas, M. Sassenburg and W. A. Smith, J. Mater. Chem. A, 2015, 3, 19556–19562 RSC.
- J. Luo, D. A. Vermaas, D. Bi, A. Hagfeldt, W. A. Smith and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600100 CrossRef.
- M. B. McDonald, S. Ardo, N. S. Lewis and M. S. Freund, ChemSusChem, 2014, 7, 3021–3027 CrossRef CAS PubMed.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS PubMed.
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L.-T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, ACS Catal., 2018, 8, 6560–6570 CrossRef CAS.
- P. Lobaccaro, M. R. Singh, E. L. Clark, Y. Kwon, A. T. Bell and J. W. Ager, Phys. Chem. Chem. Phys., 2016, 18, 26777–26785 RSC.
- J. Jung, Y. S. Jeong, U. Lee, Y. Lim and C. Han, Ind. Eng. Chem. Res., 2015, 54, 3865–3878 CrossRef CAS.
- I. M. A. Fonseca, J. P. B. Almeida and H. C. Fachada, J. Chem. Thermodyn., 2007, 39, 1407–1411 CrossRef CAS.
- N. Schüler, K. Hecht, M. Kraut and R. Dittmeyer, J. Chem. Eng. Data, 2012, 57, 2304–2308 CrossRef.
- K. M. Guthrie, Chem. Eng., 1970, 24, 114–142 Search PubMed.
- W. G. Sullivan, E. M. Wicks and J. Luxhoj, Engineering Economy, Prentice Hall, 2003 Search PubMed.
- J. M. Douglas, Conceptual Design of Chemical Processes, McGraw-Hill, 1988 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00647h |
| This journal is © The Royal Society of Chemistry 2020 |