
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroelectrochemistry, the future of visualizing electrode processes by hyphenating electrochemistry with spectroscopic techniques
Jasper J. A.
Lozeman†
 ,
Pascal
Führer†
,
Wouter
Olthuis
and
Mathieu
Odijk
*
,
Pascal
Führer†
,
Wouter
Olthuis
and
Mathieu
Odijk
*
BIOS Lab-on-a-Chip Group, MESA+ Institute, University of Twente, 7522 NB Enschede, The Netherlands. E-mail: m.odijk@utwente.nl
First published on 15th January 2020
Abstract
The combination of electrochemistry and spectroscopy, known as spectroelectrochemistry (SEC), is an already established approach. By combining these two techniques, the relevance of the data obtained is greater than what it would be when using them independently. A number of review papers have been published on this subject, mostly written for experts in the field and focused on recent advances. In this review, written for both the novice in the field and the more experienced reader, the focus is not on the past but on the future. The scope is narrowed down to four techniques the authors claim to have the most potential for the future, namely: infrared spectroelectrochemistry (IR-SEC), Raman spectroelectrochemistry (Raman-SEC), nuclear magnetic resonance spectroelectrochemistry (NMR-SEC) and, perhaps slightly more controversial but certainly promising, electrochemistry mass-spectrometry (EC-MS).
 Jasper J. A. Lozeman | Jasper Jeroen André Lozeman graduated for his BSc in Analytical Chemistry in 2013 at the HU University of Applied Sciences Utrecht (the Netherlands). In 2015 he completed his MSc in Chemistry, Analytical Sciences at the Vrije Universiteit Amsterdam (the Netherlands). Currently, he is a PhD researcher at the BIOS Lab-on-a-Chip group of the MESA+ Institute of Nanotechnology, at the University of Twente (the Netherlands). His current project is titled “surface enhanced vibrational spectroscopy in a flow-through microfluidic chip” funded by the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC). |
 Pascal Führer | Pascal Führer finished his cooperative studies in Chemistry and Biotechnology at the Niederrhein University of Applied Sciences in Krefeld, Germany, in 2015 with a BSc. degree. He then studied Chemistry at the University of Münster, Germany, with a focus on analytical and inorganic chemistry, obtaining an MSc degree in 2017 while investigating the synthesis and reactivity of amido-substituted siliconoid clusters. Currently, he is developing a new generation of microfluidic chips for the spectroelectrochemical investigation of human drug metabolism mimicry and protein cleavage as a PhD researcher in the BIOS Lab-on-a-Chip group at the University of Twente in the Netherlands. |
 Wouter Olthuis | Wouter Olthuis received his MSc. degree in electrical engineering from the University of Twente, Enschede, the Netherlands. Then, he joined the Center for MicroElectronics, Enschede (CME). After that, he started his PhD project and received his doctoral degree in 1990. Since 1991 he has been working as an Assistant Professor in the Laboratory of Biosensors of the University of Twente on physical and (bio)chemical sensors. Currently, he is Associate Professor in the BIOS Lab-on-Chip group of the MESA+ Institute of Nanotechnology. He has (co-)authored over 180 papers (h = 40) and 7 patents. From 2006 until 2011 he has also been the Director of the Educational Programme of Electrical Engineering at the University of Twente. |
 Mathieu Odijk | Mathieu Odijk (1981) is a professor leading research in Microdevices for Chemical Analysis, as part of the BIOS Lab-on-a-Chip group at the University of Twente. His research team currently consists of 10 PhD students and 1 post-doc, with a shared aim to push existing boundaries in analytics using micro- and nanofabricated devices. He received an MSc degree in electrical engineering in 2007, and a PhD in electrochemical microreactors in 2011. He has been nominated as technological top talent in 2012, and awarded a prestigious personal VENI award in 2014 by the Dutch research council (NWO). |
1 Introduction
1.1 Spectroelectrochemistry (SEC)
Spectroelectrochemistry is an established technique which hyphenates electrochemistry with spectroscopy. Electrochemistry by itself is a technique that can be used in order to determine concentrations of known compounds or to obtain information concerning reaction kinetics. However, it is less suitable for elucidating unknown reaction intermediates or products.1 By combining electrochemistry with an optical technique, more qualitative and quantitative information about the processes occurring at the electrodes can be obtained.It is generally accepted in the SEC field that the work of Kuwana et al.2 in 1964 is the first true SEC experiment. This early work on spectroelectrochemistry has resulted in a field containing a large variety of spectroscopic methods. Nowadays, a number of reviews exist concerning spectroelectrochemistry. For example the work by Dunsch from 2011, covering a wide range of multi-spectroelectrochemistry techniques.3 In 2013, Oberacher et al. published a paper on mass spectrometric methods in electrochemical cells.4 Wain and O'Connel wrote a paper in 2017 about surface-enhanced vibrational spectroelectrochemistry.5 Also in 2017, work by Tong on nuclear magnetic resonance spectroelectrochemistry focused on the challenges and prospects was published.6 The work by León and Mozo, published in 2018, describes in detail how to design a spectroelectrochemical cell.7 In 2018, Zhai et al. wrote a review in which they describe the recent advances in spectroelectrochemistry.8 Finally, in the work by Gazor-Ruiz et al. from 2019, the recent trends and challenges of spectroelectrochemistry are described.9
1.2 Visualizing the future of SEC
Most of the papers mentioned above are addressed towards experts in the fields, with a strong focus on recent advancements. This review tries to add to these existing review papers, firstly by focussing on the future of spectroelectrochemistry and secondly by writing a review paper in an accessible way for newcomers to the field. To be concise, the current review is focused on the four techniques that, in the opinion of the authors, have the biggest potential to undergo major improvements in the coming decades. The techniques covered in this review are infrared spectroelectrochemistry (IR-SEC), Raman spectroelectrochemistry (Raman-SEC), nuclear magnetic resonance spectroelectrochemistry (NMR-SEC) and, perhaps slightly more controversial but certainly promising, electrochemistry mass-spectrometry (EC-MS). These techniques will each be discussed in their own sections in the aforementioned order. The basic principle of every technique is first explained, followed by the current state of the art in the field. To conclude, every chapter ends with a future perspective, based on the developments in the separate fields of the SEC technique and the substantiated opinion of the authors. The authors hope that this review will inform both the newcomers as well as the experts concerning the future of SEC. At best, we aim to give an overview of how the future of SEC may look like and at worst, to initiate a scientific discussion on the subject.2 Infrared spectroelectrochemistry (IR-SEC)
2.1 Introduction IR-SEC
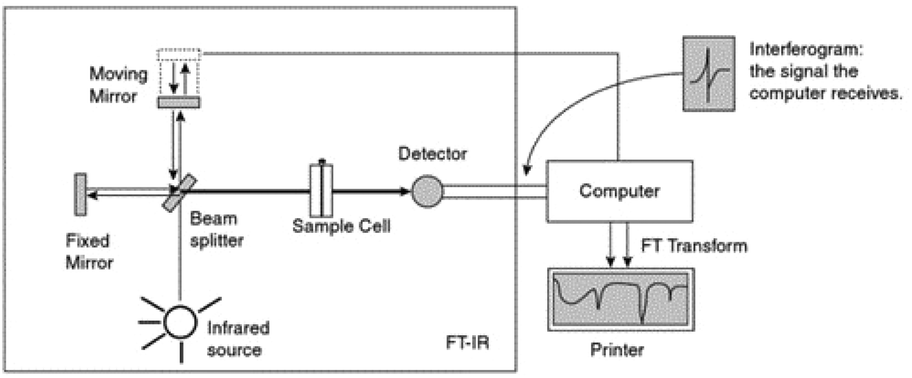 | ||
| Fig. 1 Schematic diagram by Pavia et al.10 of an FTIR instrument. From Pavia/Lampman/Kriz/Vyvyan. Introduction to Spectroscopy, 4E. © 2009 Brooks/Cole, a part of Cengage, Inc. Reproduced by permission. http://www.cengage.com/permissions. | ||
When considering IR spectroscopy, there are some key drawbacks associated with the technique. Most importantly, IR instruments often use a silicon carbide rod (such as a Globar), heated to 1000 °C or above, as a light source. Although these sources can cover a large spectral window, their power output is relatively low. This results in a relatively high detection limit compared to other analytical techniques. Another serious drawback is that water has strong absorbance bands in the IR region, which complicates measuring in aqueous solutions. In order to prevent the water bands from dominating the spectrum, Lambert–Beer law (eqn (1)) offers a practical solution.
| A = ε·c·l | (1) |
Due to the requirement of such a small path length, an often used cell design in IR-SEC is the so called “thin-layer” configuration. This configuration is used as follows: the sample with electrolyte is applied on an electrode and a window of IR-transparent material (such as CaF2) is pushed against the electrode. As a result the layer of liquid analyte is sandwiched between the electrode and the transparent IR window, with a thickness of several μm. A drawback of this method is that the exact thickness of the thin-layer is not as reproducible as with a fixed cell size, making the process of taking a background reference spectrum more difficult. The thickness of the layer can be calculated by measuring the absorbance of the bulk water vibration,13 potassium ferricyanide,14 or any other substance with a known concentration and molar attenuation coefficient and applying the Lambert–Beer law. This determination of the path length is not necessarily done using IR spectroscopy. De Lacey et al.15 used UV/Vis spectroscopy of 8 mM cytochrome c to determine the path length of their thin-layer cell. Once the path length of both the sample and the background measurement is known, a background correction can be performed.
An alternative way to make a background correction when using the thin-layer configuration is by difference spectroscopy. In difference spectroscopy, a background reference measurement is performed at one potential and then subtracted from the measurement taken at a different potential. In this way, the contribution of the bulk solution is cancelled out and a spectrum is produced showing only the changes caused by the variation of the potential. There are, however, drawbacks to this technique, as adsorption and desorption of the analyte on the electrode changes the concentration of the measured analyte.16
2.2 State of the art of IR-SEC
The design by Moss et al.11 is shown in Fig. 2. As illustrated, a working electrode (WE) in the form of a Au mesh is sandwiched between two IR-transparent windows (CaF2), creating a thin-layer configuration. A Teflon body keeps the cell together. The width of the cell and the WE is in the same order of magnitude as the width of the IR-light beam, allowing for the analysis of the entire cell. The design by Moss et al.11 has been used by several researchers over the past few years with little change in the design.15,28–31,33 Most changes are small, such as changing the WE material into a platinum mesh instead of a Au mesh.30,31,33 One notable setup that does not use the above-mentioned mesh configuration is the work done in the group of Swain,34–36 where boron doped diamond (BDD) deposited on undoped silicon is used as transparent electrode material. This has as benefit over the mesh configuration that light is not blocked by the electrode. Among the large number of applications found in literature, some common applications for transmission IR-SEC are investigations of proteins, such as their oxidation, reduction and folding properties.11,15,28,29 Another application is the study of porphyrins.30,31,33
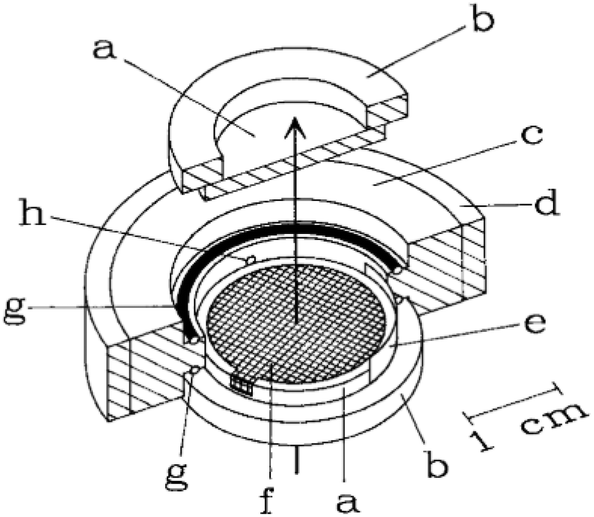 | ||
| Fig. 2 Schematic representation of the IR-SEC cell. The arrow represents the propagation of the IR-light beam (a) IR-transparent CaF2 window mounted onto a (b) Plexiglas ring, (c) Teflon body, (d) steel surround, (e) Pt counter electrode, (f) the mesh working electrode, (g) O-ring, (h) capillary connection to the reference electrode. Reprinted from the original work by Moss et al.,11 Copyright 1990, with permission from John Wiley and Sons. | ||
2.2.2.1 Basic principles reflection IR-SEC. The basic principles for IR reflection spectroscopy are as follows: the IR light is emitted by the light source. The IR light propagates through an IR-transparent window (often CaF2) into the electrochemical cell. Next the light beam hits the electrode, an IR reflective material, and is reflected out of the cell, as shown in Fig. 3. The reflected beam passes back through the same window into the detector. In Fig. 3 a typical schematic representation of a reflectance cell by Alwis et al.14,37 is shown. The WE is on top of a CaF2 window creating a thin-layer configuration. The reference electrode (RE) is separated from the cell by a Luggin capillary and the counter electrode (CE) is looped around the WE.
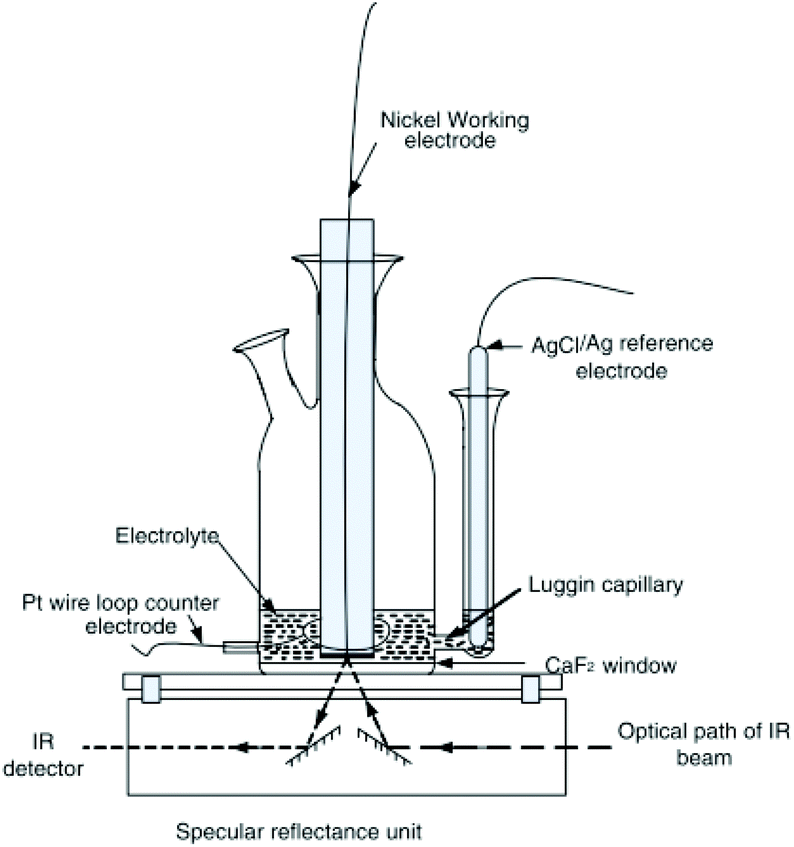 | ||
| Fig. 3 Schematic representation of a reflection setup. Republished with permission of Journal of The Electrochemical Society, original by Alwis et al.,14 Copyright 2013; permission conveyed through Copyright Clearance Center, Inc. | ||
Drawbacks of reflectance IR-SEC compared to transmittance IR-SEC mostly come from the somewhat more complex setup. When using a reflection setup it is necessary to introduce extra mirrors into the IR-spectrometer in order to focus the incident beam onto the electrode and focus the reflected beam onto the detector. Aligning these mirrors can be a time-consuming process, more labour intensive compared to transmission spectroscopy. The use of extra mirrors also introduces extra losses due to part of the light being absorbed instead of being reflected. An advantage of using reflection compared to transmission spectroscopy is that no light is blocked by the electrode material.
2.2.2.2 Fibre optic reflection IR-SEC. An alternative for the standard reflection setup is fibre optic reflectance spectroscopy. First developed by Shaw et al.38 and used over the past decade primarily in, or in collaboration with, the group of Richter-Addo.38–41 The advantage of this specific configuration compared to reflection systems such as those by Alwis et al.14,37 is the ease of alignment of the source with the sample and electrode, allowing for smaller configuration of the setup. With the small thin-layer setup they claim better control over the applied potential than transmittance and ATR cells.38 This particular configuration is mostly applied to the study of porphyrins,39–41 although it should be noted that it can also be used for applications where reflection IR is currently used.
2.2.2.3 SNIFTIRS and PM-IRRAS. As mentioned in section 2.1.2, IR-SEC often operates with a thin-layer configuration. In reflection mode there are two different methods in order to perform background corrections. The first method, based on difference spectroscopy and most often reported in literature, is subtractive normalized interfacial Fourier transform infrared reflection spectroscopy (SNIFTIRS). Alternatively, polarization modulation infrared reflection absorption spectroscopy (PM-IRRAS) is used. SNIFTIRS is an IR-SEC reflection technique first introduced in the early 1980s by Pons.42 The authors would like to note that the use of the term SNIFTIRS seems to be used inconsistently throughout literature. Several papers use the acronym SNIFTIRS,14,37,42–44 while other papers, seemingly using SNIFTIRS, don't make use of the term.13,45–50 SNIFITRS is a technique used for a number of different applications, such as the study of the oxidation of small organic molecules (such as ethanol and ethylene glycol)13,45,48,49 and metal ions.14,37,43 SNIFTIRS is also performed in combination with SEIRAS, for example for the studies of proteins.46,47
PM-IRRAS, an alternative for SNIFTIRS, was first performed by Golden et al.51 in 1981. After this initial work, further notable improvements on the technique were done by Faguy et al.,52,53 the group of Corn54,55 and the group of Lipkowski.56–59 PM-IRRAS is most often used in cases where the potential modulation necessary for SNIFTIRS could damage the analyte of interest.16 Additional advantages of PM-IRRAS over SNIFTIRS, as proven by Faguy et al.,52,53 include that PM-IRRAS is very insensitive to atmospheric water and carbon species and has an increased signal to noise ratio.59,60 PM-IRRAS is especially suitable for measurements of samples adsorbed on metal surfaces, such as electrodes. Explained in a simplified manner, in PM-IRRAS the polarization of the reflected light is modulated and the reflection takes place under a grazing angle of incidence. Under s-polarization (perpendicular to the plain of incidence) there is no interaction between the light and electrode surface. However, when using p-polarization (parallel to the plain of incidence), constructive interference takes place at the electrode surface. By cycling the polarization of the light, a difference spectrum of the bulk (s-polarization) and at the electrode surface (p-polarization) can be obtained. PM-IRRAS has been reported in literature as, among others, a method for the study of self-assembled monolayers61,62 and lipid bilayers.56–59
For a more in-depth description of SNIFTIRS and PM-IRRAS the authors recommend reading the work of Guidelli,16 Bard63 and Alkire et al.59
2.2.2.4 Electrode material and SEIRAS in reflection IR-SEC. Concerning SEIRAS in reflection IR-SEC, electrode materials used are Ni,14,37,43 Pt,13,38–41,50,64 Pd45,48,49 and Au.46,47,62,65,66 While only Liu et al. report on using the reflecting electrodes for surface enhanchement,46,47 it is unlikely that the Au electrodes used by Hosseini et al.,62 Su et al.65 and Villalba et al.66 don't report any enhancement. For Pt, Pd and Ni, SEIRAS has been reported in the past,67 but none of the papers above mentioned FTIR-SEC papers report this effect. This might be because it is not relevant for their application or that the SEIRAS effect is negligible.
2.2.3.1 Basic principles of ATR-IR-SEC. Attenuated total reflection infrared spectroscopy (ATR-IR) operates under the following principle: IR light is coupled into an internal reflection element (IRE), as shown in Fig. 4. When the light hits the sample interface at an angle higher than the critical angle (θcrit), total internal reflection occurs, reflecting the light and creating an evanescence wave. This evanescence wave penetrates into the sample for several micrometres, with the electric field decaying at an exponential rate away from the surface. This has the advantage that the effective path length in ATR-IR is always in the order of several μm, and thus removes the need for a thin-layer configuration. In order to get a good estimation of the penetration depth in ATR-IR-SEC the following formula can be used:68
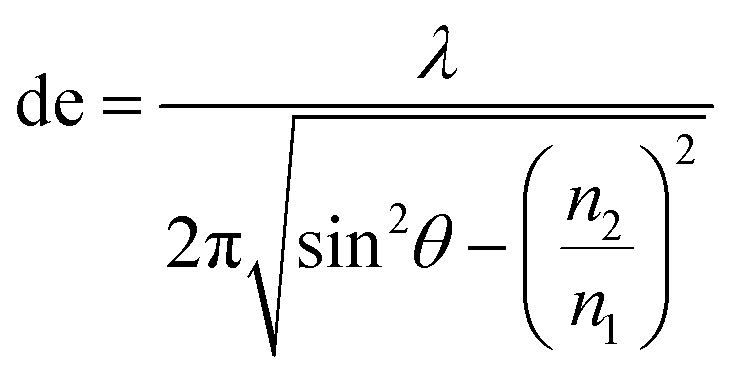 | (2) |
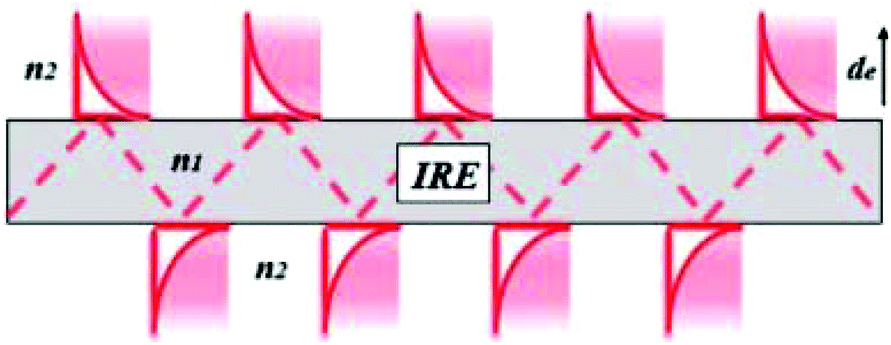 | ||
| Fig. 4 Internal reflection element. Reprinted and adapted from the original work by Schadle and Mizaikoff,68 Copyright 2016, with permission from SAGE Publications. | ||
In this equation de is defined as the penetration depth (m) at which the intensity of the wave is reduced to 1/e of its original value, or approximately 37%. Furthermore λ is the wavelength (m) used, n1 is the refractive index of the IRE, n2 the refractive index of the sample and θ is the angle of incidence (°).68 This formula can give a good estimation of the penetration depth and the effect of the IRE material on the penetration depth of the system. It can consequently be used to get an estimate of the path length through the sample.
2.2.3.2 IRE material ATR-IR-SEC. Penetration depth is one reason to choose for a certain ATR material, other reasons are availability, hardness and inertness. In Table 1, three different materials used for the IRE in ATR-IR-SEC are shown: diamond, ZnSe and Si. Diamond is more expensive than ZnSe or Si, but its large spectral window68,69 (2–25 μm), its chemical inertness and the high hardness are clear advantages. ZnSe is cheaper than diamond and has a similar spectral window68,69 (1–25 μm) but is relatively soft and can only be used between pH 5–9. Finally, and also cheaper than diamond, there is Si which is harder than ZnSe and somewhat more inert, but suffers from a reduced spectral window68,69 (2–7 μm). Surprisingly, Ge, a commonly used ATR material, does not seem to be used in ATR-IR-SEC. A possible reason for this is the fact that Ge is more conductive than diamond, ZnSe and Si and therefore could potentially cause interference with the simultaneous operation of the electrochemical cell.
| IRE | Setup | Electrode material | Configuration | Author |
|---|---|---|---|---|
| Diamond | III | Carbon nanofibres | Single bounce | Dillard et al.77 |
| Diamond | I | Boron doped diamond | Single bounce | Neubauer et al.82 |
| Diamond | II | Boron doped diamond/Au nanoparticles | Single bounce | Izquierdo et al.81 |
| López-Lorente et al.317 | ||||
| Si | III | Carbon paper | Multi bounce | Healy et al.76 |
| Si | III | Carbon paper | Multi bounce | Paengnakorn et al.71 |
| Si | II | Au/Pd | Not mentioned | Yang48 |
| Si | II | Au | Not mentioned | Cheuquepán et al.318 |
| Si | II | Au | Single bounce | Dunwell et al.319 |
| Si | II | Cu/Au | Single bounce | Heyes et al.78 |
| Si | II | Au | Single bounce | Ataka et al.21 |
| ZnSe | I or II | Sn/SnOx | Single bounce | Baruch et al.74 |
| ZnSe | I | Nitrogen doped diamond like carbon | Multi bounce | Menegazzo et al.83 |
| ZnSe | I | Au | Single bounce | Pfaffeneder-Kmen et al.79 |
| ZnSe | I | Boron doped silicon | Single bounce | Purushothaman et al.70 |
| ZnSe | II | Au | Single bounce | Viinikanoja et al.80 |
| ZnSe | II | Au | Multi bounce | Zimmermann et al.320 |
Another variable in Table 1 is the configuration of the beam path through the IRE. As shown in the table, a single bounce or a multi bounce configuration can be used. Reasons to choose a certain configuration include the law of Lambert-Beer (eqn (1)), since a multi bounce configuration, where the light interacts more often with the sample, effectively has a longer path length.
2.2.3.3 Electrode material and configuration in ATR IR-SEC. Also reported in Table 1 are the three different setups for the placement of the WE. The first configuration (denoted as I in Table 1), is the so called optically transparent electrode. This is either made out of a thick Au mesh deposited on the IRE, similar as those reported in transmission IR-SEC (2.2.1), or an optically transparent layer deposited on top of the ATR crystal. These optically transparent electrodes, such as boron doped diamond (BDD), nitrogen doped diamond like carbon (N-DLC) or boron doped silicon (BDSi), could potentially also be used as ATR material, but this does not seem to be applied in practice. A possible reason for this could be fabrication limitations. A schematic representation of an ATR-IR-SEC design using the optically transparent electrode configuration can be seen in Fig. 5.
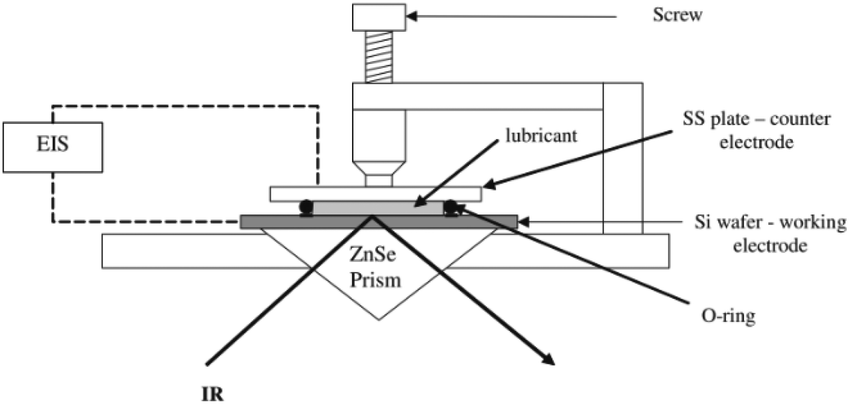 | ||
| Fig. 5 Schematic of an optically transparent electrode ATR IR-SEC setup, in a single bounce configuration. Reprinted with permission from Springer Nature: from the original work by Purushothaman et al.,70 Copyright 2012. | ||
The second configuration (denoted as II in Table 1), is similar to the first configuration in the sense that the electrode material is deposited on the top of the IRE, but instead of an optically transparent material or mesh, a thin layer of metal is deposited. This setup, known as the Kretschmann configuration,72,73 is a method in ATR FTIR to perform SEIRAS experiments. The thin metal layer here acts as an electrode and as a surface for SEIRAS simultaneously. This thin-layer electrode has some drawbacks: if the layer is too thick, too much IR absorbance by the material occurs, if it is too thin, holes in the continuous layer might occur. Baruch et al.74 deposited a thin layer of Sn/SnOx as electrode material on top of an ATR crystal. They do not mention any SEIRAS effects, but one might argue that it is likely that SEIRAS does take place. The reasoning for this is that Sn has been reported to have surface enhanced properties in the infrared region,75 making it debatable whether it should be in configuration I or II.
The final configuration (denoted as III in Table 1) uses a thin porous spacer between the IRE and the electrode. As one can see in Table 1 and in Fig. 6, a thin porous layer, either a Nafion membrane,71 carbon nanoparticles76 or carbon fibres,77 is placed on top of the IRE. A carbon paper electrode is placed in contact with this layer, covering the surface and functioning as a WE. This setup has as advantage that it does not need labour intensive deposition techniques.
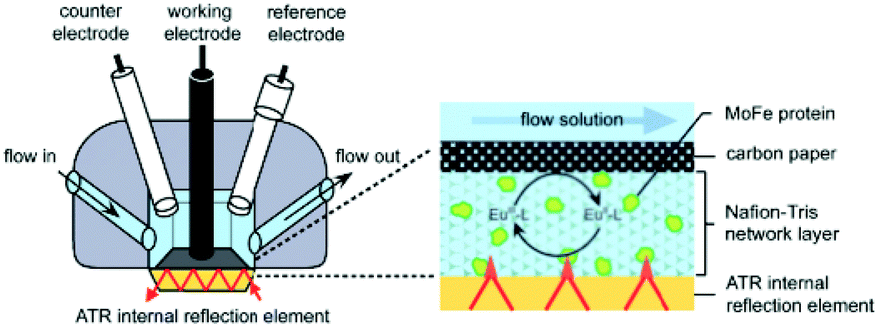 | ||
| Fig. 6 Schematic representation of the cell configuration using a spacer between the IRE device and the electrode. As shown, the Pt CE and calomel RE have more space than in the thin-layer configuration used for transmission and reflectance IR-SEC. In the inset one can see the IRE, the porous Nafion membrane containing the analyte and the carbon paper electrode on top. A multi-bounce configuration is used. Reprinted from the original work by – Paengnakorn et al.,71 Copyright 2017, published by The Royal Society of Chemistry. | ||
A large number of applications regarding ATR-IR-SEC are reported in literature. Some examples are the study of metalloproteins,71,76 the study of CO2 reduction without74 and with the combination of SEIRAS,78 the reduction of graphene oxide,79,80 technique development81–83 and practical applications such as observing lubricant degradation.70
2.2.4.1 Conventional source. The most notable work in using IR-SEC spectroscopy with a conventional source comes from the group of Sun,84 who developed their own IR-SEC microscope in the late 1990s. They further developed this instrument in the last two decades for a variety of different applications regarding electrode interface reactions.85–87 Sun and Zhou also wrote a paper in which they further explain the use of FTIR microscopy for electrochemical applications.88
2.2.4.2 Synchrotron source. Next to IR microscopy using conventional IR sources, instruments using synchrotron radiation have also been reported. The first experiments regarding the use of synchrotron radiation with electrochemistry were reported by Melendres et al.89 in 1995, although the IR frequency used was in the far infrared. The first experiment that coupled mid infrared synchrotron radiation with electrochemistry was the work by the group of Burgess90 in 2011 where they proved that, when using small aperture settings, synchrotron radiation has a better signal to noise ratio than conventional sources. Further notable work with synchrotron IR microscopy combined with electrochemistry was done by the group of Burgess on spatial mapping of methanol oxidation on a PtNi alloy,91 femtomole detection of ferrocyanide on ultra-microelectrodes92 and quantitative analysis of electrochemical diffusion layers.93 Other notable work regarding synchrotron radiation in combination with electrochemistry was done by Ash et al.94 on synchrotron based infrared microscopy to study biological redox processes under electrochemical control. In this paper they show that they can observe changes in the Flavin mononucleotide active site of a flavoenzyme with synchrotron radiation.
Although the power of synchrotron radiation is proven by the work done by researchers mentioned above, it has to be noted that the need of synchrotron radiation is a major drawback to using synchrotron based IR spectroscopy. Access to a synchrotron source is limited and will stay so in the foreseeable future.
2.3 Future perspective
The fields of IR and SEC are ever increasingly combined and advanced. Recently, a three-electrode electrochemical ATR cell from Pike Technologies called the Jackfish SEC Cell has appeared on the market. The J1W version of this device uses commercially available disposable IRUBIS ATR-SEIRAS devices, making IR-SEC suitable for a broader audience.95It can be postulated that further improvements in the field of IR-SEC are linked to the advancements made in the fields of IR spectroscopy and electrochemistry. These advancements will take some time to be implemented into IR-SEC, but it is logical to assume that any advancement improving the separate fields, will eventually be implemented in IR-SEC.
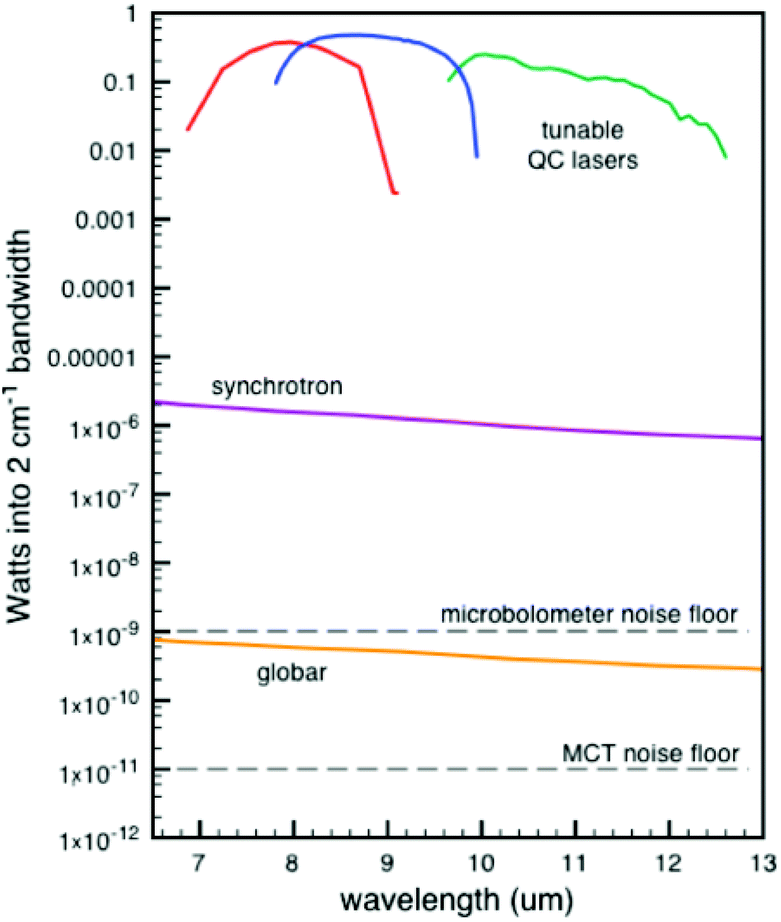 | ||
| Fig. 7 Comparison of Weida and Yee of three different IR sources of the signal through a 10 μm pinhole.96 Reprinted and adapted from the original work by Weida and Yee,96 Copyright 2011, with permission from SPIE and M. Weida. | ||
The comparison as shown in Fig. 7 is made when passing the light through a 10 μm pinhole. Subsequent spot sizes of the three techniques are normalized. This comparison highlights the strength of QCLs in IR-microscopes, yet for normal spectroscopy this graph is skewed somewhat in favour of the QCL device. Nonetheless, the main message stays the same, being that a higher source power can be obtained with QCLs. Due to the higher power output of the QCLs, a shorter measurement time compared to regular IR instruments can be achieved. A commercially available QCL system from IRsweep, using optical heterodyne detection, even further improves this time resolution. This IRsweep instrument can take a spectrum with a spectral width in the order of 100 cm−1 within 4 μs. This allows for a more accurate understanding of the dynamic processes occurring at the electrode interface.
A drawback of QCLs is the shorter wavelength coverage compared with standard IR sources. The wavelength range of a single QCL is usually somewhere in the order of a micrometre. This can be compensated to some extent by having several lasers in a system. QCLs have already been used for several IR techniques,97,98 and therefore could be implemented in IR-SEC in the near future.
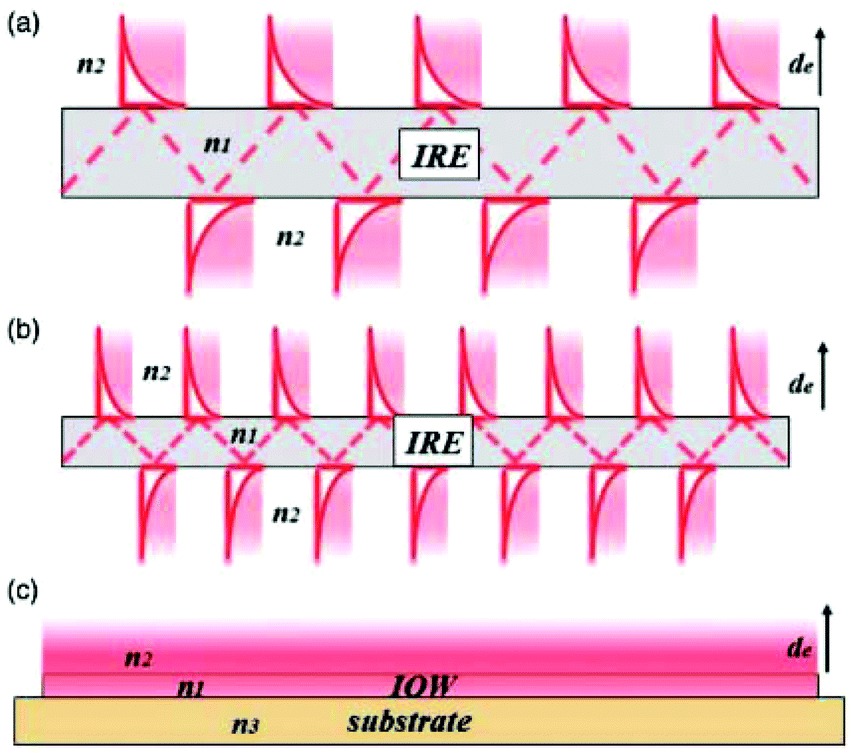 | ||
| Fig. 8 Schematic representation of the evanescent wave/field with an ever decreasing IRE thickness. As one can see in (a) and (b), the smaller IRE has more reflections than the bigger IRE. When the size is small enough, as one can see in (c), a waveguide is created with a continuous evanescent field. Reprinted from the original work by Schadle and Mizaikoff,68 Copyright 2016, with permission from SAGE Publications. | ||
When the already small waveguide also functions as electrode, a device with a small footprint can be created. This way it can be easily combined with microfluidics in order to make lab-on-a-chip systems.
Although less established than electrochemistry, the combination of IR with microfluidics is also a developing field, for example: an ATR-IR device for the study of electric field-driven processes,109 reaction monitoring110 and the analysis of chemical reaction intermediates.111 A recent review written on spectroscopic microreactors for heterogeneous catalysis by Rizkin et al. also includes a section about IR.112
The combination of IR-SEC and microfluidics is not an established field yet, although some work has been done by Führer et al.113 It is expected that in the near future more work regarding IR-SEC is published.
2.3.5.1 PTIR. PTIR is a technique that combines AFM with infrared spectroscopy. This hyphenated technique has the optical resolution of the AFM technique and the ability to obtain the chemical information usually obtained with IR. In PTIR, the probe tip of an AFM microscope is positioned close to the region of interest. Next, a tuneable IR laser is focused on the region close to the probe tip. If the sample has a vibrational frequency that corresponds to the wavelength emitted by the laser, absorption will take place. This absorption results in a thermal expansion of the sample, which then will be measured by the AFM cantilever.114,115 The resulting spatial resolutions for commercially available instruments are in the order of 20 nm, at least two orders of magnitude lower compared to optical microscopy.114,115 To our knowledge, this technique has not yet been combined with SEC. One possible explanation could be that SEC introduces too much heat in the sample, which could interfere with the AFM measurement.
2.3.5.2 Nano-FTIR. In Nano-FTIR, FTIR and scattering-type scanning near-field optical microscopy (s-SNOM) are combined. IR light from a conventional IR laser source or a synchrotron source is focused on a conductive AFM probe tip. This probe tip acts as an antenna for the IR light and focuses and enhances it. The near field interaction between the probe tip and the sample influences how the light is being scattered. Recording the scattered light results in a spectrum of the sample. While imaging the surface, a map with special resolution as small as 10 nm can be obtained.114,116,117 One application of Nano-FTIR combined with SEC was reported by Lu et al.116 where they study the molecular structure of graphene-liquid interfaces, allowing new possibilities for the characterization of graphene-electrolyte interfaces.
3 Raman spectroelectrochemistry (Raman-SEC)
3.1 Introduction Raman-SEC
Raman is often considered to be complementary to IR spectroscopy. The reason for this is that, in order to be visible in Raman spectroscopy, the vibration has to experience a change in polarizability. In other words, a change in how easy it is to distort the electron cloud around the molecule. As mentioned in the previous section, IR active molecules require a change in dipole moment in order to be visible in IR spectroscopy. Often vibrations that are polarizable do not have a dipole moment and vice versa, making the techniques complementary.
A typical setup used in Raman spectroscopy is shown in Fig. 9, as reported by Schmid and Dariz.119 It consists of a light source, most often a laser, focused on the sample using a microscope objective. The scattered light is guided back into the instrument and a filter removes the Rayleigh scattered light. The Raman scattered light is guided onto the grating of a monochromator and eventually reaches the detector, where it records the resulting spectrum.
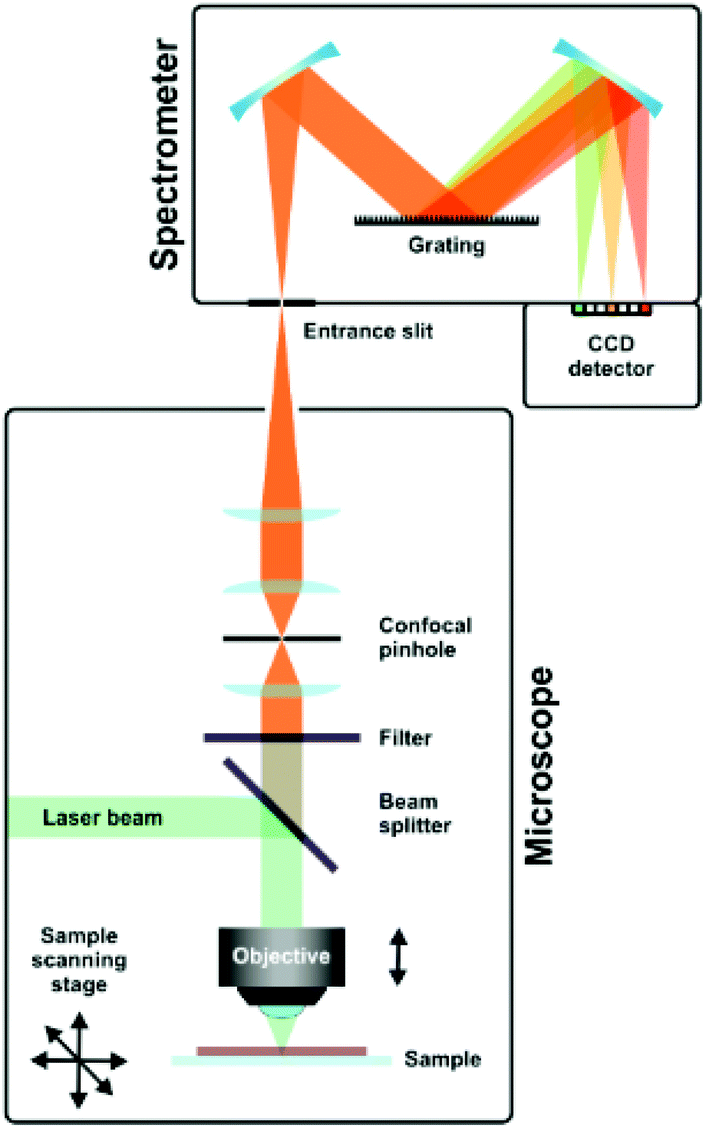 | ||
| Fig. 9 Schematic representation of a Raman setup. Reprinted from the original work by Schmid and Dariz,119 Copyright 2019, with permission from Creative Commons. Licensed under CC BY 4.0. | ||
A second factor for enhancement that should be discussed is the “chemical” enhancement. Unlike the well-defined electrochemical enhancement, the term “chemical” enhancement describes more of a collection of different phenomena.127 Simplified, the chemical enhancement mostly describes phenomena due to the charge transfers between the analyte molecule and the metal substrate and other metal substrate/analyte molecule interactions. The chemical enhancement is significantly lower than the electrochemical enhancement, and much harder to theoretically explain and predict.126
In practice, distinctions can be made between three different kinds of SERS substrate. The first are randomly orientated enhancement surfaces, such as roughened (electrode) surfaces and randomly orientated nanoparticles deposited or grown on a surface, which are relatively easy to fabricate. Second are the so called structured arrays, which can exist of a self-assembled array nanoparticles deposited on a substrate or nanofabricated arrays. These structured arrays can obtain higher enhancement factors than the roughened surfaces but are more difficult to fabricate. Finally, in order to improve measuring in liquid samples, metallic particles can be used in order to create SERS active colloidal solutions.24 This last form, using colloidal particles, cannot be used in SERS-SEC, since it cannot function as or be in contact with the WE easily. With the right combination of SERS substrate material and configuration of the structures, enhancement factors of up to 1010 can be achieved, depending on the used definition of SERS enhancement.24
One important note on working with nanoparticles is the need for proper cleaning of the particles in order to maximize the SERS effect. The recent work of Montiel et al. describes some of the methods that can be used for the cleaning of these particles.128
SERS is a complex field with persistent misconceptions.124 Therefore a complete review on the subject is beyond the scope of this study. The authors of this review recommend that the reader reads the work cited for a more in-depth explanation of SERS.24,67,120–126,129
3.2 State of the art of Raman-SEC
In the literature, several different cell configurations are described. Most commonly used is the stationary electrode configuration. Other configurations include the rotating cell, the linearly moving cell or the flow through cell. Two important reasons to choose a certain cell are the ease of use and the balance of fabrication versus potential photo-degradation. Photo-degradation of samples can be a severe problem in Raman spectroscopy. Constant illumination of a substrate can heat up the sample and eventually cause burning or evaporation of the sample or matrix. In the next section, we will discuss the different cells, their advantages and disadvantages, and how they deal with photo degradation.
3.2.1.1 Stationary electrode. The stationary electrode, as shown in Fig. 10 is the most commonly used configuration. Since the laser is stationary with respect to the electrode, alternatives should be considered in order to reduce damage due to the laser beam. Depending on how susceptible the sample is to photo degradation, one should limit the laser power and the exposure time of the sample.
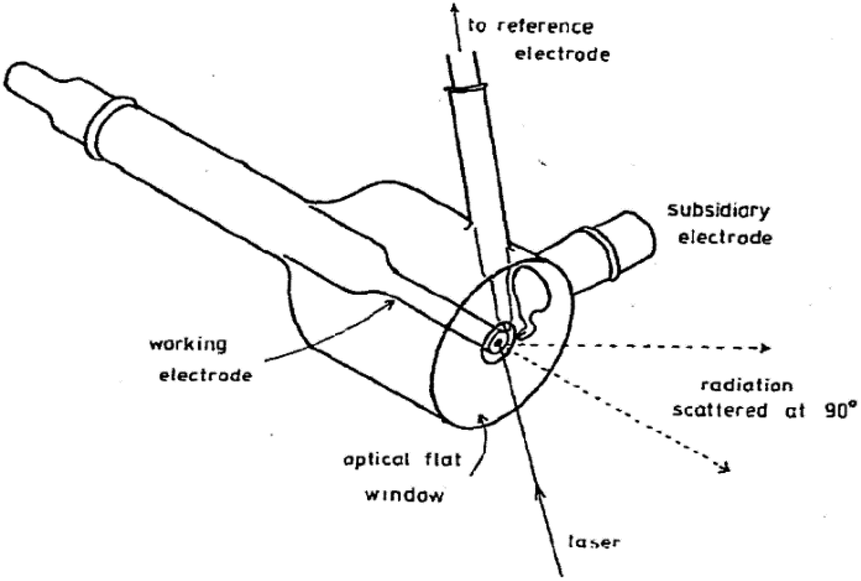 | ||
| Fig. 10 Example of a stationary electrochemical cell used for Raman experiments. Reprinted from the original work by Fleischmann et al.,120 Copyright 1974, with permission from Elsevier. | ||
3.2.1.2 Linearly moving electrodes and cells. In order to reduce photo degradation, several setups have been designed to limit the time the Raman laser remains on a single spot on the substrate. One such a device operates by moving the entire cell and its electrode as can be seen in Fig. 11. The cell moves together with the electrode in a linear path with respect to the Raman light source, reducing the time for which a single spot on the electrode is exposed to the Raman source. The linear moving electrode was first developed by Niaura et al.140 and is mostly used by Mažeikienė et al. to study polyaniline,141–146 dyes147–150 and hexacyanoferrates.151,152
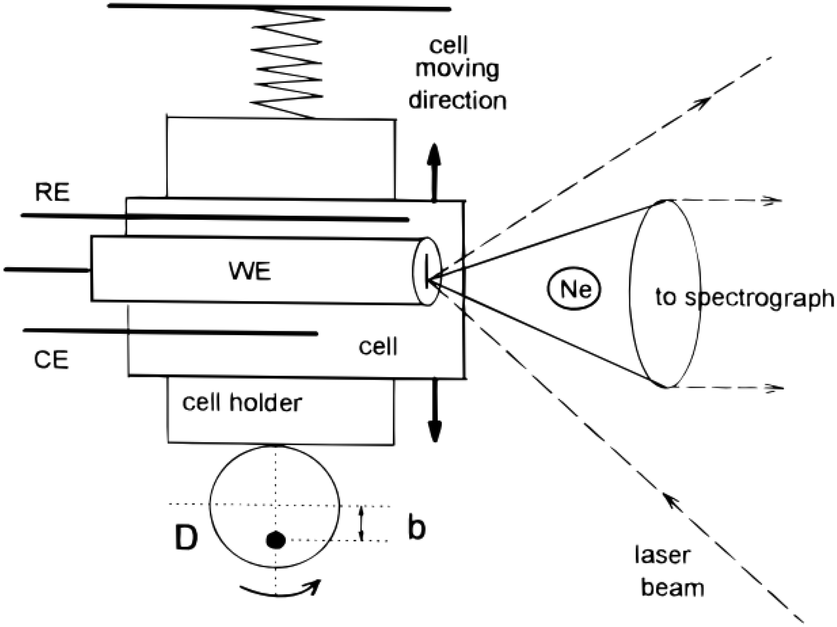 | ||
| Fig. 11 Schematic of a moving electrode cell. Ne is the Neon lamp and D is the rotating disk rotating around the axis represented by the black dot. Reprinted from the original work by Niaura et al.,140 Copyright 1997, with permission from John Wiley and Sons. | ||
3.2.1.3 Rotating electrodes and cells. Next to the moving electrode system, a rotating Raman system has been first developed by Kiefer and Bernstein153 in 1971. One of the first rotating Raman-SEC setups was reported by Yamada et al.154 in 1978, as shown in Fig. 12. An advantage from an electrochemical point of view for both the rotating electrode and the moving electrode is that there are no mass transport limitations due to the added convection. Some use of the rotating electrode has been reported in the 70's and 80's,154–158 but, despite the potential of the rotating electrode, little mention of its use is reported in recent literature. An alternative to the rotating electrode configuration is the rotating cell configuration, in which instead of the electrode the entire cell is being rotated. This device was first used by Kavan et al.159 in 2009. The WE is positioned at the centre of the motor axis while the sample rotates. Despite the potential for this setup, it is only mentioned once more in literature, for Fullerene C60 in an aqueous medium.160
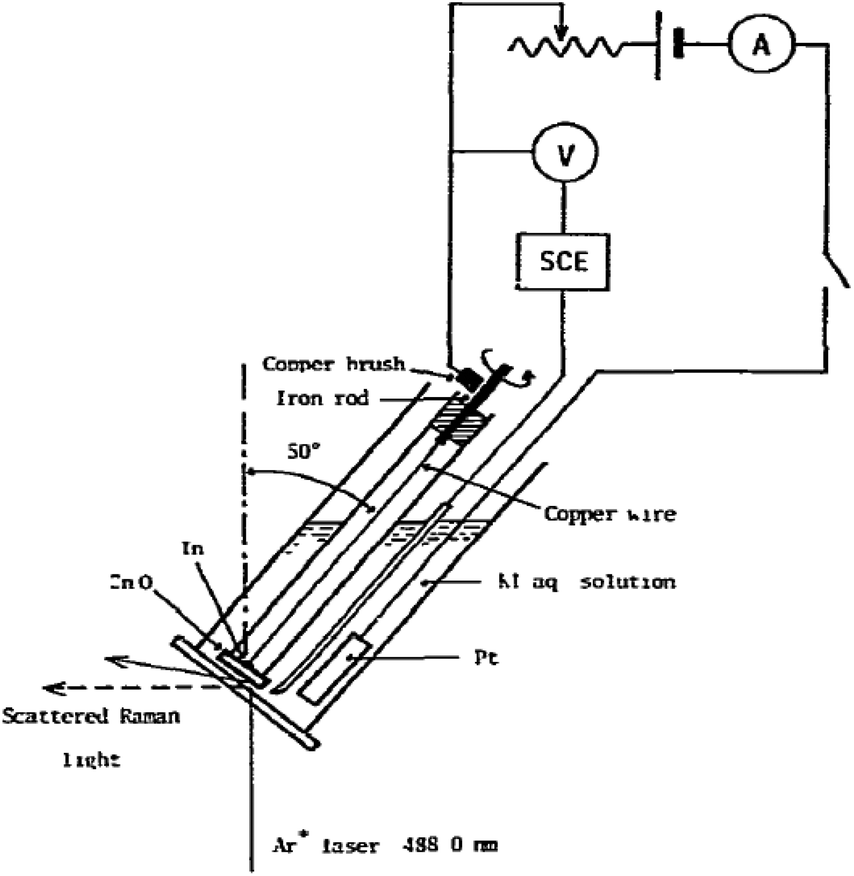 | ||
| Fig. 12 Rotating Raman-SEC device. Reprinted from the original work by Yamada et al.,154 Copyright 1978, with permission from Elsevier. | ||
3.2.1.4 Flow through cell. Another way to reduce the chance of photo-degradation is a flow through cell, which is easy to use for non-viscous liquids, but not applicable for solid samples. Most commonly, a flow through cell either operates by having a constant flow of fresh liquid flowing over the WE161,162 or by continuous mixing of the electrolyte solution.163
3.2.1.5 Alternative designs. A special configuration that is worth mentioning is the setup by Ibañez et al.,164 using a 4-electrode system in order to study the charge transfer at a polarisable liquid/liquid interface. Another interesting design is the Raman spectroscopy of a carbon super capacitor by Bonhomme et al.165
3.2.2.1 Materials. Raman-SEC is used in a multitude of research fields and for different applications. An important research field for Raman-SEC is material science to investigate novel materials. One example is the research done on carbon nanotubes. Most of this extensive research comes from the groups of Dunsch and Kavan,166–177 although some work has been performed elsewhere.178–183 A related field is the investigation of graphene.162,163,184–190 Another popular application of Raman-SEC within material science is to get more insight on conductive polymers, studied by the group of Malinauskas141–146,191 and others.192–202 A number of applications are reported on the investigation of dyes,147–150,203,204 small organic molecules that can form monolayers on the electrode material205–209 and protein/cell studies.140,210–212
3.2.2.2 Sensing applications and point of care. Research aimed towards real world applications is also present in Raman-SEC, although underrepresented and almost solely performed by the group of Brosseau. Some interesting applications include a portable SERS-SEC device for the detection of melanine in milk,136 the detection of DNA biomarkers in synthetic urine,137 the detection of 6–thiouric acid in synthetic urine,213 the quantitative detection of uric acid in synthetic urine138 and a point of care device based on SERS-SEC on a fabric substrate.139
3.3 Future perspective
Raman-SEC is already a more mature technique than for example IR-SEC. This does not mean, however, that the technique has stopped to progress. The authors of this review are of opinion that most future development will take place on the SERS substrates and the development of new applications for Raman-SEC, especially towards real world applications.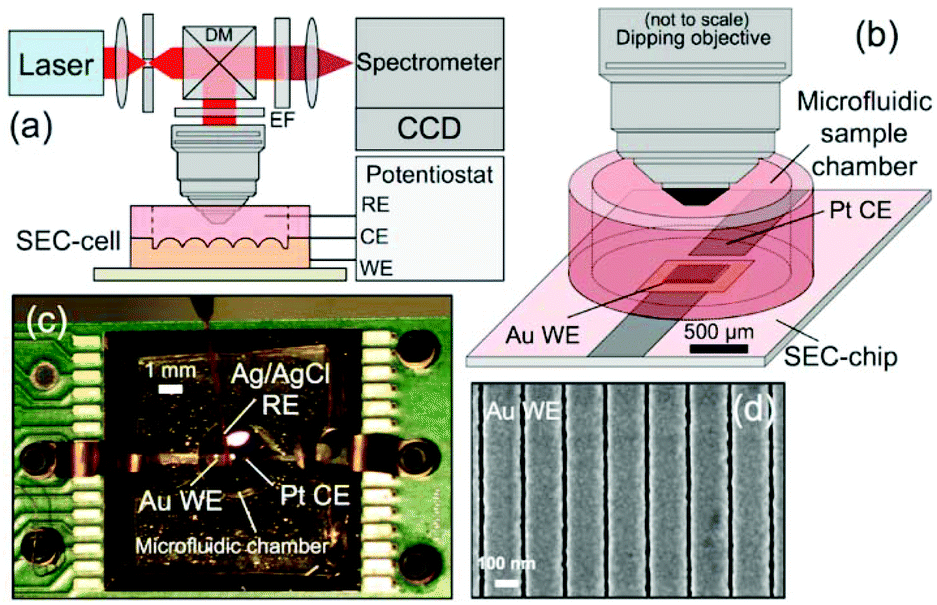 | ||
| Fig. 13 In situ SERS-SEC system by Yuan et al.214 (a) Schematic representation of the Raman SEC analysis system, (b) SEC-cell composed of a SEC-chip directly bonded to a small-volume microfluidic sample chamber with an optical interface to a microscope objective, (c) SEC-cell consisting of a Pt CE and nanostructured Au WE patterned on the Si SEC-chip and an external Ag/AgCl RE, and (d) representative SEM image of the Nanostructures Au WE. Reprinted with permission from Yuan et al.214 Copyright 2015 American Chemical Society. | ||
Compared to, for instance, Au islands and randomly distributed particles, these nanostructured arrays can achieve a higher reliability, since the enhancement is higher and equal over the entire surface. This makes these structures more suitable for quantitative measurements compared to the randomly organized substrates.
3.3.3.1 Time resolved Raman spectroscopy. Most commonly, Raman-SEC is performed using the so-called steady state method. In the steady state method, the electrode potential is set to a desired value and given time to stabilize.219,220 After this stabilization period, a Raman spectrum is acquired. This steady state method has the benefit of being highly reproducible in obtaining data. However, information is lost when operating in this steady state configuration, such as information on reactive intermediates. Several experiments have been done in the 1990s regarding time resolved Raman spectroscopy.221–226 Van den Beld et al. have recently used an in-house developed graphene spectroelectrochemical cell to study the adsorption behaviour of simple redox couples in a more dynamic fashion, i.e. acquiring real-time Raman spectra while recording cyclic voltammograms at a scan rate of 25 mV s−1.190 Also recently, Zong et al.220 published on a transient SERS-SEC instrument that can acquire data at millisecond time resolution without the need to repetitively cycle the data acquisition. Further improvements in the field of time-resolved Raman-SEC might lead to a better understanding of reaction intermediates at the electrode surface.
3.3.3.2 Spatial resolution. One method in Raman spectroscopy that can be used in order to improve the spatial resolution is tip-enhanced Raman spectroscopy (TERS). First proposed by Wessel227 in 1985 and first independently developed by Stöckle et al.,228 Anderson,229 Hayazawa et al.230 and Pettinger231 in 2000. In short, a metal or metal-coated tip is illuminated with a laser. The metal tip acts as a substrate for surface enhancement and can therefore be used for sensing purposes. In general, TERS achieves an enhancement factor of about 103 to 106, and has a spatial resolution between 10–80 nm,232 much smaller than the diffraction limit of light. The combination of SEC and TERS is a relatively new field, first introduced in 2015 by Kurouski et al.233 and Zeng et al.234 As an emerging field, we expect that TERS-SEC will find more interest in the future.
4 Nuclear magnetic resonance spectroelectrochemistry (NMR-SEC)
4.1 Introduction NMR
Nuclear magnetic resonance (NMR) spectroscopy is a technique which is often used to obtain structural data about analytes. It typically operates within radio frequencies in the range of 60 to 1000 MHz (wavelengths of ca. 5 to 0.3 m). These low-energy waves can interact with nuclei that possess a magnetic spin, for example the isotopes 1H and 13C. For NMR spectroscopy, the different spin states of the nuclei get separated with a powerful magnetic field. The surrounding atoms and functional groups in a molecule influence how strongly the outside magnetic field affects the target nucleus locally. As a consequence, NMR spectroscopy is able to obtain comprehensive structural information of the molecule.The sample can be manipulated and analysed with radio frequency radiation generated with a transmitter inside the spectrometer and the NMR probe around the sample. Nowadays, there are many different pulse patterns being used for sample excitation under varying experimental conditions.
For more detailed information, the authors refer the reader to a recent review paper by Colnago et al.237 and the references therein, explaining the fundamentals and challenges of NMR spectroscopy.
4.2 State of the art of NMR-SEC
NMR spectroscopy is a versatile tool to identify molecules non-destructively and accurately. It is most commonly used for the analysis of organic compounds, although there are also many applications which involve metals or metalorganic compounds. This versatility makes NMR spectroscopy a good coupling partner to EC, which can e.g. struggle to attribute signals correctly in the presence of side reactions. By combining the two techniques, however, mechanistic and kinetic information can be obtained in situ accurately and in a much more flexible way than e.g. with UV/Vis or IR spectroscopy, which can be limited by the functional groups present in the analyte.However, because of the strong dependency of NMR measurements on the electromagnetic environment of a nucleus and the involvement of a strong outside magnetic field, NMR-SEC is not trivial. Combining EC with NMR leads to interference between the electrical and the magnetic fields, often caused by the electrode configuration, which can worsen the signal to noise ratio and signal resolutions in general. Additionally, instrumental failure upon simultaneous use of both systems has been reported, making real-time in situ measurements challenging.238 Pioneering work on NMR-SEC was performed by Wieckowski and associates. Many of their reported approaches were based on transferring the reaction products from an EC-cell into an NMR tube and then analysing them with an NMR spectrometer in an offline manner.239–249 Other works included dedicated SEC-NMR cells250,251 or placing an EC-cell into an NMR spectrometer.252
A simple way to avoid possible interference between the two methods is to use an online technique which separates the electrochemical and the NMR-spectroscopic steps. However, this leads to much longer response times than is necessary to observe electrochemical intermediates and short-lived reaction products and is difficult to automate. To our knowledge, online EC-NMR methods are rarely being reported.
In this review paper, we instead focus on the liquid sample in situ EC-NMR methods reported in recent years and briefly mention their respective applications. For information on NMR applications in battery research, Hu et al. have recently published a review paper.254
4.2.1.1 Carbon fibre electrodes. Recent publications using carbon fibre WEs are based on a design originally published by Klod et al. in 2009.253 The design is shown in Fig. 14 and incorporated three electrodes. The WE and CE both consisted of carbon fibre filaments in sealed glass capillaries and the pseudo-RE was a PTFE-covered Ag wire with a AgCl tip. Only the active region of the WE was placed in the NMR detection area. The motivation to use carbon fibres was the higher possible reaction rate to obtain enough electrolysis product for 1H and 13C NMR measurements in lower amounts of time. Recent applications of this cell included the analysis of p-benzoquinone,253,257 2-diphenylaminothiophene based compounds258 and a perfluoroalkylated fullerene.259 In the last case, 19F NMR spectroscopy was performed as opposed to the more common 1H and 13C experiments. The measurements in all of the applications just mentioned were performed over large timeframes of up to 17 hours of electrolysis with periodic NMR spectroscopy sampling. Nunes et al. used a very similar configuration with the RE placed above the NMR detection area in a 10 mm NMR tube for the reduction of 9-chloroanthracene.238 They used a steady-state free precession (SSFP) pulse sequence to obtain a strong enough signal for 13C experiments in the span of 11 minutes while halting the 150 minutes long electrolysis process during the measurement. Huang et al. used a different variation of the three-electrode design in which the WE carbon fibres just above the NMR detection area were loaded with a commercial electrocatalyst and a Pt gauze served as CE.260 They reported successful 13C measurements of the electrocatalytic oxidation of 13C-labeled ethanol by averaging 128 scans.
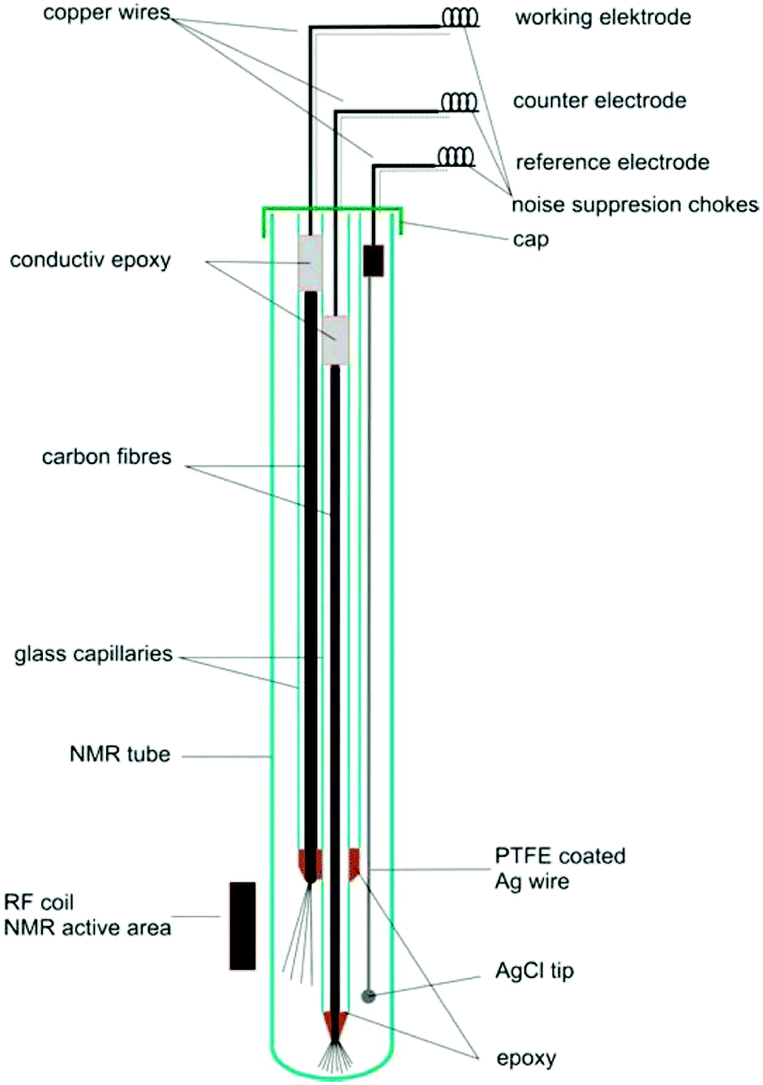 | ||
| Fig. 14 Schematic of a three-electrode NMR-SEC cell with carbon fibre WE and CE, partially placed in the NMR detection area. Reprinted with permission from Klod et al.253 Copyright 2009 American Chemical Society. | ||
In 2013, Bussy et al. reported a slightly different three-electrode configuration in which carbon microfibres were again used as WE and CE.261 The RE was replaced with a Pd wire. In this iteration, shown in Fig. 15, the electrodes were interfaced with the glass capillaries slightly differently and placed in different positions. The CE was still placed beneath the NMR detection area while the WE and the pseudo-RE were placed above it. Using this design, the oxidation of phenacetin was monitored periodically over the span of 6 hours.261 Another application of this NMR-SEC cell was reported by Boisseau et al. when they performed ultrafast 2D COSY experiments on 9-chloroanthracene.262 Periodic NMR samples were taken every 3 minutes in a chronoamperometric experiment over a timeframe of 80 minutes.
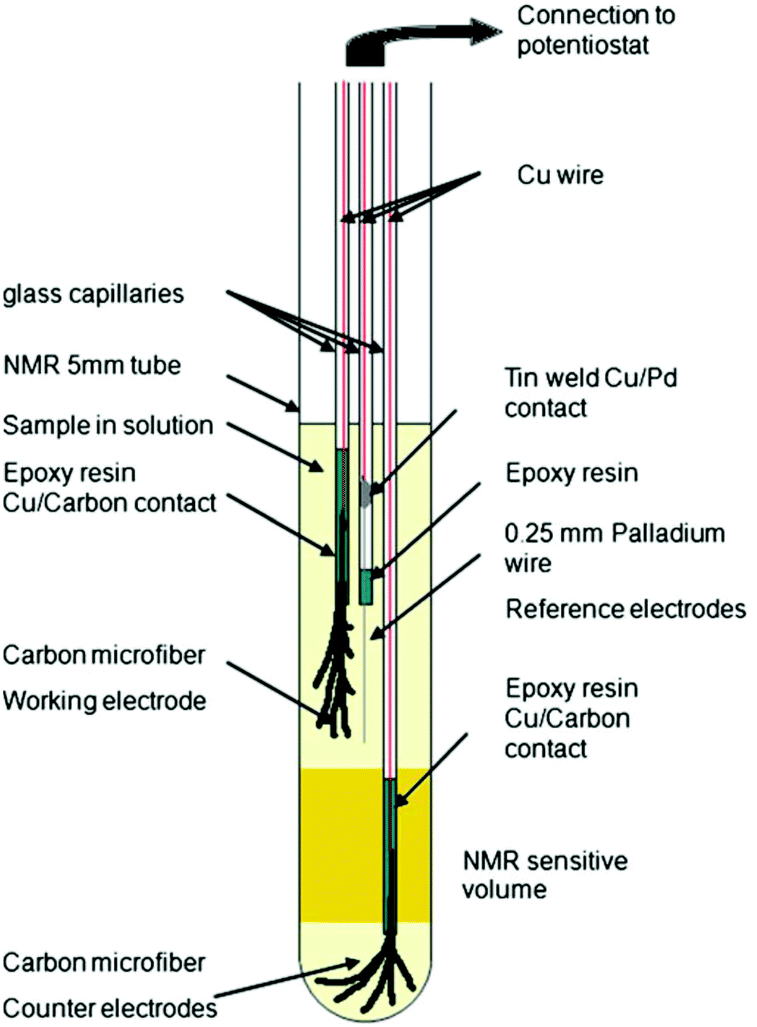 | ||
| Fig. 15 Schematic of a three-electrode NMR-SEC cell with carbon microfibre WE and CE placed above and below the NMR detection area. Reprinted by permission from Springer Nature: from the original work by Bussy et al.,261 Copyright 2012. | ||
While the previously described cell designs focused on placing the WE close to or in the NMR detection area Ferreira Gomes et al. reported an NMR-SEC cell with three-electrode setup where all electrodes were deliberately placed above the NMR detection area. They used a carbon fibre WE, a platinized Pt CE, and a Ag/AgCl pseudo-RE.263 By moving the electrodes outside of the detection area, Ferreira Gomes et al. investigated the influence of the interference between the magnetic and the electric fields on mass transport within the cell. They reported that, by exploiting the resulting magnetoelectrolysis effect,264–266 which causes the solution to be stirred by a magnetohydrodynamic force, the electrochemical reaction rate for the electrochemical reduction of p-benzoquinone was greatly increased. The reaction was monitored via1H NMR spectroscopy.
4.2.1.2 Thin film electrodes. While carbon microfibre approaches could aim to improve the electrochemical conversion inside the NMR-SEC cell, thin films with large surface areas hold the advantage of being near transparent to radio frequency radiation if they are thinner than their respective skin depth.267 This makes it possible to move the WE and possibly CE further into the NMR detection area to better monitor the electrochemical reaction close to the electrodes. One of the older approaches to introduce electrodes into an NMR tube based on a metal film was published by Zhang and Zwanziger in 2011.256 They used a thin Au film deposited on a 3 mm glass tube with small bored holes for ion exchange that served as WE and extended into the NMR detection area. A pseudo-RE was positioned inside of the WE glass tube and a curved platinized Pt-foil CE was placed between the NMR tube wall and the WE. With this metal film-based setup, Zhang and Zwanziger investigated the spectroelectrochemistry of p-benzoquinone, caffeic acid and 9-chloroanthracene in mechanistic approaches and with sampling times of up to two minutes. Cao et al. reported a similar NMR-SEC cell in which a Au coated glass tube sealed with a Nafion membrane to allow ion-exchange was used.268 Additionally, a Pt wire CE was placed inside the WE tube and a Pt foil pseudo-RE was placed between the NMR tube wall and the WE tube. Only the WE reached into the NMR detection area. They performed real-time 1H NMR-SEC in situ measurements on quinone.
In more recent publications, Sorte et al. constructed an NMR-SEC cell with an interdigitated Au WE and CE (IGEs) in the NMR detection area and a Ag/AgCl pseudo-RE above it.269,270 The design is shown schematically and as a photograph in Fig. 16. The IGEs were printed on polyimide films and could be brought into close proximity inside a 5 mm NMR tube to promote better electrochemical performance. Sorte et al. successfully used this setup for in situ NMR-SEC measurements of the electrochemistry of ferrocene (1H)269 and of the oxidation of methanol and ethanol (13C).269,270
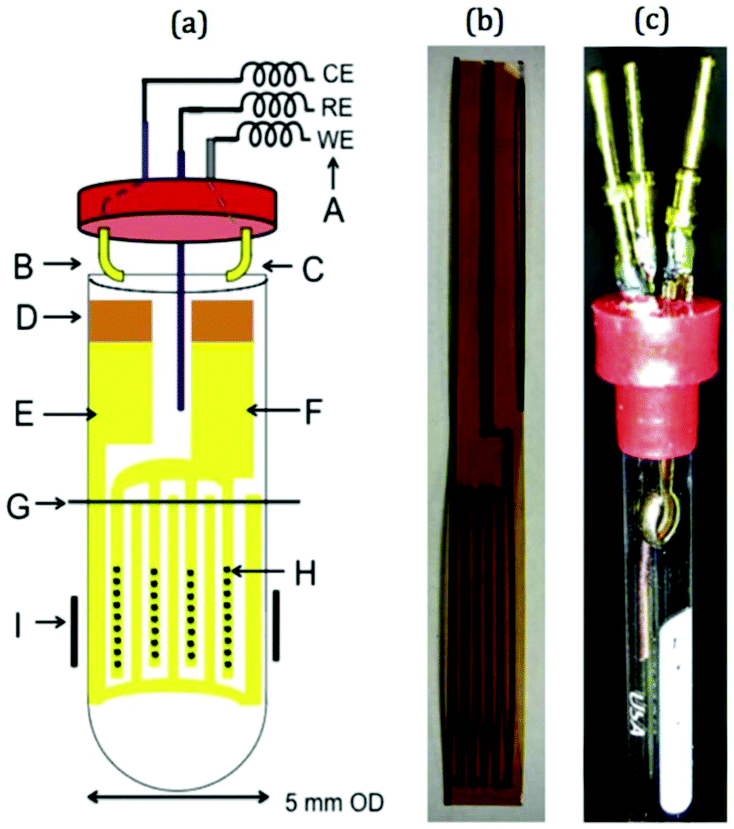 | ||
| Fig. 16 Schematic and photograph of an NMR-SEC cell with interdigitated WE and CE placed in the NMR detection area. Reprinted from the original work by Sorte et al.,269 Copyright 2016, with permission from Elsevier. | ||
Different modified tin oxide glass based coatings were explored by Zhang et al., Wang et al. and Cao et al.271–274 Zhang et al. created a nano-polyaniline film (PAn) via electro-polymerization on an indium tin oxide (ITO) electrode.271 The schematic and a photograph are shown in Fig. 17. This PAn WE was then assembled with a Pt wire CE and a Ag pseudo-RE inside of a 5 mm NMR tube where only the WE extended into the NMR detection area. Using this simple NMR-SEC, they conducted real-time 1H NMR-SEC experiments with hydroquinone. In 2019, Wang et al. used the same cell design to characterize the electrocatalytic performance of composite catalysts consisting of varying combinations of Pt, carbon, MoS2 and graphene nanosheets electrodeposited on ITO-based WEs.272,273 They investigated the oxidation of ethanol with real time 1H NMR-SEC measurements, among other techniques.
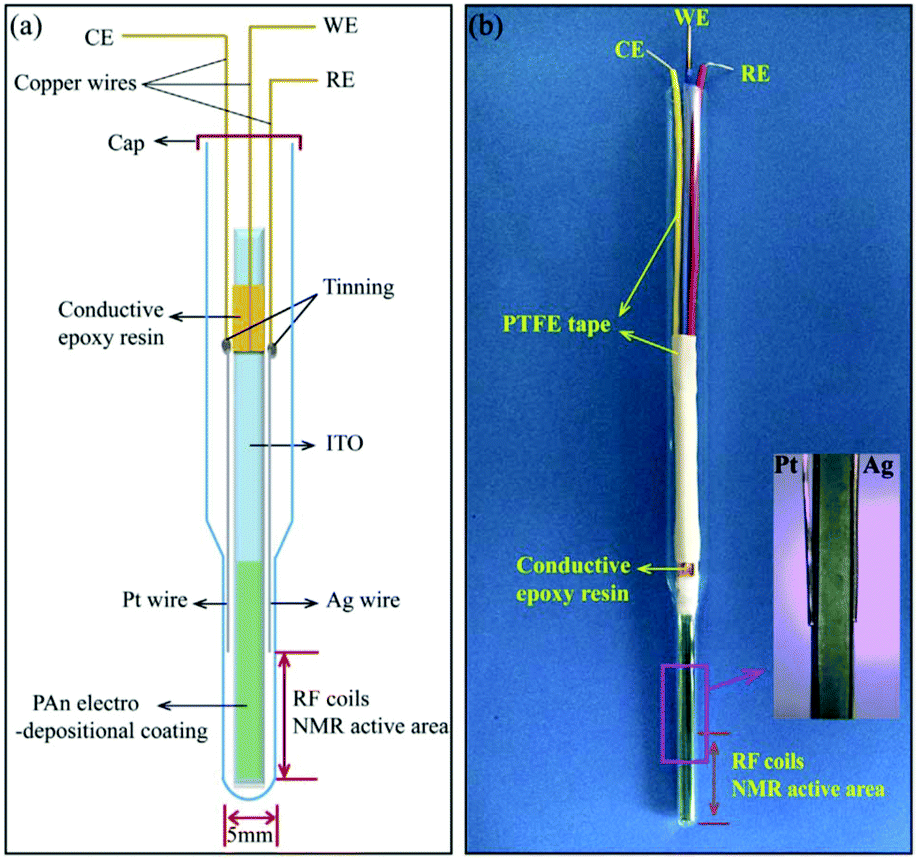 | ||
| Fig. 17 Schematic and photograph of an ITO/PAn WE based three-electrode NMR-SEC cell. Reprinted from the original work by Zhang et al.,271 Copyright 2018, with permission from Elsevier. | ||
Lastly, Cao et al. used a slightly different substrate with a similar cell design.274 They employed two fluorinated tin oxide glass (FTO) slices positioned back to back to create a two sided WE and used a Pd wire as pseudo-RE. After synthesizing Pt nanocrystals on the WE surface, they characterized the electrocatalytic performance of the WE to oxidize short-chain alcohols with real-time 1H NMR-SEC measurements.
4.2.1.3 Metal coil electrodes. A compromise between good NMR-SEC performance and a simple fabrication process was reported by da Silva et al. in 2019.275 They focused on developing a clutter-free NMR-SEC cell with simple materials and provided an instructional video on how to assemble such a cell. The design schematic is shown in Fig. 18 and comprises two Pt wire coils around a glass capillary as WE and CE, as well as a Ag wire inside the capillary as pseudo-RE. All components were located above the NMR detection area. Da Silva et al. reported using the magnetoelectrolysis effect to stir the solution in a real-time 1H NMR-SEC experiment on the oxidation of ascorbic acid, finding a doubled reaction rate in comparison to pure EC experiments.
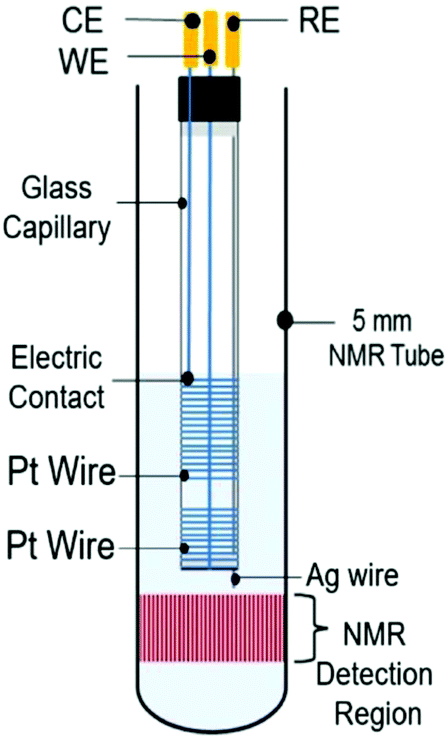 | ||
| Fig. 18 Schematic of a coil-based three-electrode NMR-SEC cell. Reprinted from the original work by da Silva et al.,275 Copyright 2019, with permission from Elsevier. | ||
The design schematic as well as photographs inside the solid state NMR spectrometer are shown in Fig. 19. With this cell and a solid state instrument, Richter et al. quantitatively monitored the oxidation of ethanol in real-time 13C NMR-SEC in situ experiments with a time-resolution in the order of 10 minutes and during a total electrolysis time of 100 hours. The cell had to be punctured due to the evolution of CO2 as reaction product.
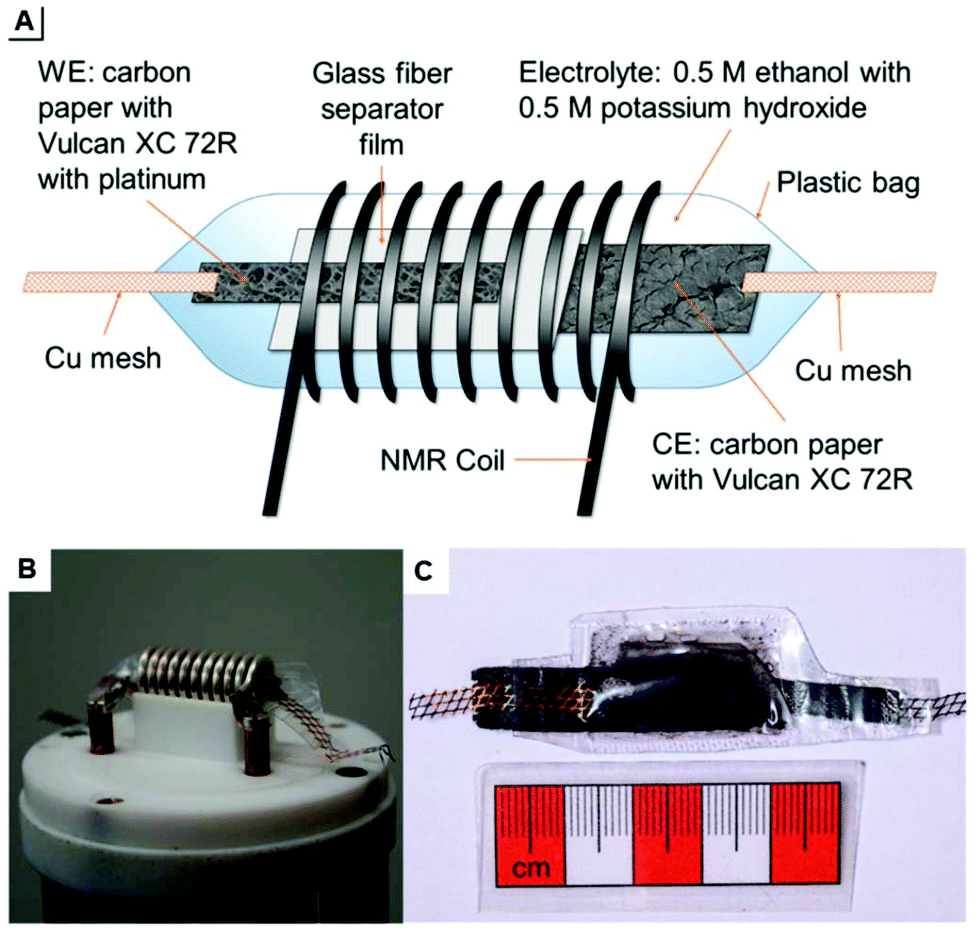 | ||
| Fig. 19 (A) schematic of a plastic pouch NMR-SEC cell and (B and C) photographs thereof. Reprinted from the original work by Richter et al.,276 Copyright 2019, with permission from Creative Commons. Licensed under CC BY 3.0. | ||
4.3 Future perspective
The integration of the EC cell into a standard NMR tube is a simple and affordable approach. Since it is always done manually, however, it may be difficult to reproduce a cell exactly, even if an instructional video is provided.275 Creating a standardized electrochemical cell insert for NMR tubes could make NMR-SEC more accessible and allow for more comparable research between different research groups and experimental setups. On the other hand, creating more specialized equipment might lead to instrumentational improvements in further avoiding negative interference between the two systems and making NMR-SEC methods more robust for real applications. An example of the influence of the instrumentation has very recently been given by Ferreira Gomes et al. in a review about the application of low magnetic field strengths for NMR-SEC.278Especially 13C NMR-SEC currently suffers from poor time-resolution. Underlying causes are low electrochemical reaction rates and the low natural occurrence of the isotope, often leading to both very long electrolysis times of tens of hours and long measurement times of tens of minutes to achieve spectra of good enough quality.
There are general NMR spectroscopy advancements such as extra sample preparation steps or specific pulse sequences that can help to improve the sensitivity and reduce the timeframe of experiments. Specific pulse techniques like e.g. SSFP are already being investigated for applications in NMR-SEC,238 but, to our knowledge, sample preparation techniques are still not being investigated. One example is dynamic nuclear polarization (DNP),6,238,254 where a solidified sample can be polarized much more strongly in the solid state and then get quickly dissolved and analysed while retaining this hyperpolarization, leading to improved signal intensities.
A different approach to increase the reaction yield and reduce the time necessary for 13C measurements could rely on the previously introduced NMR-SEC exclusive magnetoelectrolysis effect.263–266 Many cell designs rely on diffusion to move the reaction products into the NMR detection area. By investigating the exact influence of cell and electrode geometries on the stirring induced by the magnetoelectrolysis effect and optimizing them, the sensitivity limitations imposed by electrochemical reaction rates and product diffusion could be greatly alleviated. One approach to do this in the three-electrode cell type could for example focus on micromachining electrodes. By designing them in such a way that they induce defined non-uniformities in the magnetic field, the magnetoelectrolysis effect could be further investigated on a more fundamental level. Additionally, the possibilities of high-surface area electrodes to improve the conversion speed should be further investigated, since the larger currents can lead to more product but also to complications in the NMR-SEC setups.
To conclude, there are many promising areas for NMR-SEC developments to take place in the near future as it still is very much a developing technique with great potential.
5 Mass spectroelectrochemistry (EC-MS)
5.1 Introduction EC-MS
Mass spectrometry (MS) is a powerful and versatile technique to identify and quantify various kinds of analytes almost universally. The general MS process consists of the ionization of the analyte followed by the mass selection of the ions for their mass to charge ratio m/z and finally, the detection of the ions in a universal ion detector. There are many ionization and mass selector techniques with their respective advantages and drawbacks, varying for example in the ionization success for different kinds of molecules and the mass resolution that is achievable. Additionally, tandem mass spectrometry is the intentional fragmentation of the ions, for example via collision with an inert gas. The fragments can then be mass selected and detected or fragmented again for more complex experiments. More detailed information about mass spectrometry can be found in the literature.279,280While MS is not generally regarded as a spectroscopic method,281 its universal capabilities to generate qualitative as well as quantitative data make it an excellent method to pair with EC. However, three precautions need to be kept in mind: firstly, the analyte needs to be successfully ionized. Secondly, MS requires a high-vacuum environment for the mass selector. And thirdly, MS generally uses a destructive detection method, making the analyte unavailable for follow-up procedures. Because of these precautions, in situ MS measurements are not possible in the same way as with, for example, the previously described IR spectroscopy. This also means that almost no surface-related data can be obtained. However, MS is an excellent method to quantify and identify reaction products and intermediates in online approaches. The qualitative information about reaction products and their structure gained complements the electrochemical information that can be obtained, especially for more complex reactions.
In the following sections, we will present often used as well as novel and innovative experimental setups used for EC-MS measurements as well as discuss future prospects. We focus on coupling mechanisms for liquid samples and not on the electrochemical and mass spectrometric instrumentation.
5.2 State of the art of EC-MS
Liquid EC-MS can only be realized as an online method. Since the lifetime of intermediates and unstable products is limited by definition, the method needs to have a low response time between these two steps to be able to detect them. In the following sections, we present a selection of recently reported online EC-MS methods to achieve low response times and successful intermediate detection. Methods with coupled chromatography steps will be excluded since they strictly raise the response time between product generation and detection. In research questions, where no intermediates need to be detected, online EC inductively coupled plasma (ICP) MS can be used, since it destroys species information. In 2019, Kasian et al.282 reviewed EC-ICP-MS as a technique for catalyst research in an online configuration with reported response times 30–40 s.283,2845.2.1.1 Commercial thin-layer EC-MS flow cells. As has been mentioned previously,11,12 thin-layer cells and microfluidic devices can hold several advantages over bigger cell geometries for better electrochemical performance. Over the last decade, there have been many applications of commercial thin-layer flow cells for EC-MS, often but not always coupled to liquid chromatography (LC). In a review from 2014, Faber et al. covered the principal designs and flow charts of these setups,288 focusing on applications in metabolism studies. In a more recent publication, Zabel and Weber reported the use of a thin-layer flow cell setup with a response time of ∼25 s for the study of sulphuric amino acid and glutathione oxidation.289 As using thin-layer flow cells is a very established and commercialized approach, the authors do not go into further detail here.
5.2.1.2 Microfluidic EC-MS flow cells. As described previously, microfluidic EC methods can hold many advantages over methods with larger reactors. Since EC-MS methods are online techniques, a microfluidic EC cell generally does not need to be specifically designed for coupling to MS.
In 2012, Odijk et al.290 reported an EC chip coupled to a microchip-ESI, which was previously developed by Qiao et al.291 They investigated the oxidation of mitoxantrone as well as the influence of frit channels on the EC performance of the chip. As a special precaution to separate the electrical circuits, a grounded transfer tubing with high resistance was used to transfer the EC products to the microchip-ESI. Using the same chip design as Odijk et al., van den Brink et al. integrated a commercially available ESI emitter to couple the chip to an MS system287 (Fig. 20). Van den Brink had less issues with circuit separation, as the ESI needle itself was grounded while the orifice of the MS was set to high voltage. Using this latter setup, response times of ∼4.5 s were achieved.
 | ||
| Fig. 20 Photographs of a microfluidic EC cell integrated with a commercially available ESI needle in an EC-MS setup. Reprinted with permission from van den Brink et al.287 Copyright 2015 American Chemical Society. | ||
5.2.1.3 EC-MS droplet in front of MS-interface. A coupling method with a very simple configuration was employed by Yu et al. in 2018,292 as shown in Fig. 21. They used a droplet-approach in which a microscopy glass cover slide served as a carrier. This setup was developed from a previously reported pure MS approach with a glass carrier from Jiang et al.293
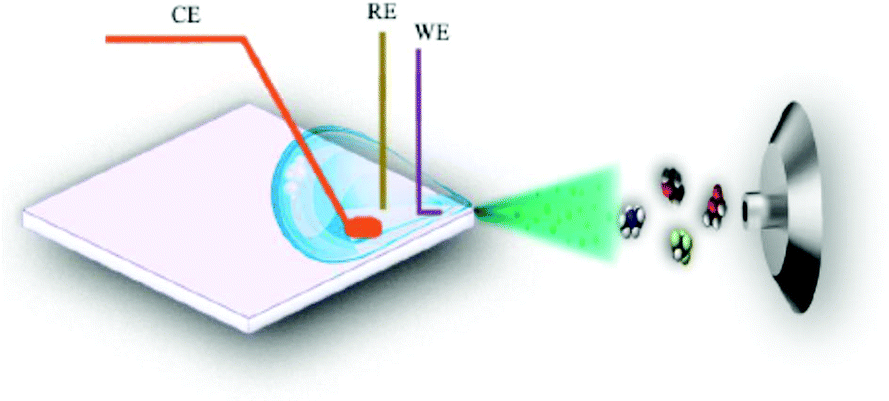 | ||
| Fig. 21 D Schematic of the droplet-based EC-MS approach. Reprinted with permission from Yu et al.292 Copyright 2018 American Chemical Society. | ||
The glass carrier was operated with a sample volume of 80 μL forming a droplet in one corner. In that corner, there was also a three-electrode setup consisting of a Pt WE and CE as well as a Ag/AgCl RE. In addition to the electrochemical reaction, the WE and CE were also used to form the electrospray together with the MS inlet. The authors do not specify a response time between the reaction and subsequent detection, but demonstrate real-time behaviour of the setup system to obtain mechanistic EC reaction data.
As a droplet-based approach, this setup is very small and simple to use. A disadvantage is the non-continuity of the experiment. The droplet is gradually desaturated of the analyte and saturated with reaction products over the course of the experiment which limits the versatility of this method. Additionally, the electrospray removes more and more liquid, slightly changing the entire system over time. The exact positioning and amount of the liquid in the droplet is also difficult to exactly reproduce. As a consequence, this setup is well suited for qualitative mechanistic studies but not for quantitative investigations.
5.2.1.4 Thin liquid layer double-barrel capillary. Qiu et al.294 and later Guo et al.295 reported a homemade capillary device where the EC reaction takes place inside a thin liquid layer formed just outside the tip of a hydrophilic double-barrel capillary. One of the two barrels is capped with previously in situ generated carbon while the other barrel contains the analyte solution as well as a AgCl/Ag wire. Primary ions for the formation of an electrospray are generated with a piezoelectric pistol at the beginning of the capillary.
By positioning the EC reaction at the tip outside of the capillary at a distance of 2 mm to the MS inlet, the response time is reduced greatly. While the authors don't give an exact value, they succeeded in detecting a diamine intermediate formed by the oxidation of uric acid with a reported half-life of about 23 ms (ref. 296) which makes this method an excellent online EC-MS technique with a very low response time. Later, Guo et al. applied this technique to detect intermediates in organometallic Ru- and Ir-based chemiluminescence experiments.
Qiu et al.294 have created a powerful online EC-MS method with a small footprint, as shown in Fig. 22. Their setup needs careful preparation to form the carbon electrode, but shows an excellent response time. The ionization process is unusual, however, and probably the most variable part between different experiments. In addition to this drawback, there is no RE, further complicating the use of this setup. With careful calibration, however, this method can serve as a great tool to investigate short-lived reaction products and intermediates.
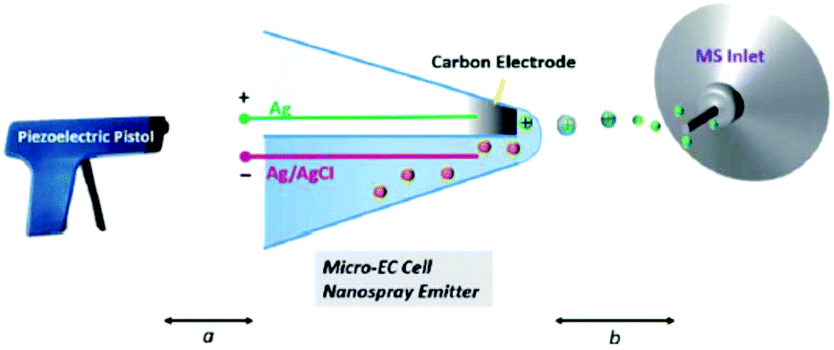 | ||
| Fig. 22 Schematic of the thin-layer double-barrel capillary setup. Reprinted from the original work by Qiu et al.,294 Copyright 2016, with permission from Creative Commons. Licensed under CC BY 3.0. | ||
5.2.2.1 Thin-layer DEMS cells. Modern DEMS systems, as described by Abd-El-Latif et al.,300 are generally built in a thin-layer configuration, where an electrode is placed on one side and a porous membrane on the other side. The electrochemical reaction products have to cross from the electrode to the other side of the cell to pass the membrane, causing response times of up to 2 s.
One example of a thin-layer cell still in use at present with modifications was published by Jusys et al. in 1999, and is depicted in Fig. 23.297 It was originally designed for a combination of DEMS and an electrochemical quartz crystal microbalance (EQCM) in two separate chambers. The membrane used in this instance is a porous Teflon membrane, placed on top of a stainless steel frit that leads to an MS inlet. In 2007, Heinen et al. slightly changed the cell to include a Pt thin-film layer on a Si prism instead of an EQCM302 to allow a combined EC-IR-MS method. These two designs have been used in multiple recent publications with applications ranging from CO oxidation and desorption on nanocrystals,303 ethylene glycol oxidation,304,305 methanol oxidation306 to the oxygen evolution reaction.307 The reported response time of the setups lies in the range of 1–2 s.297,304
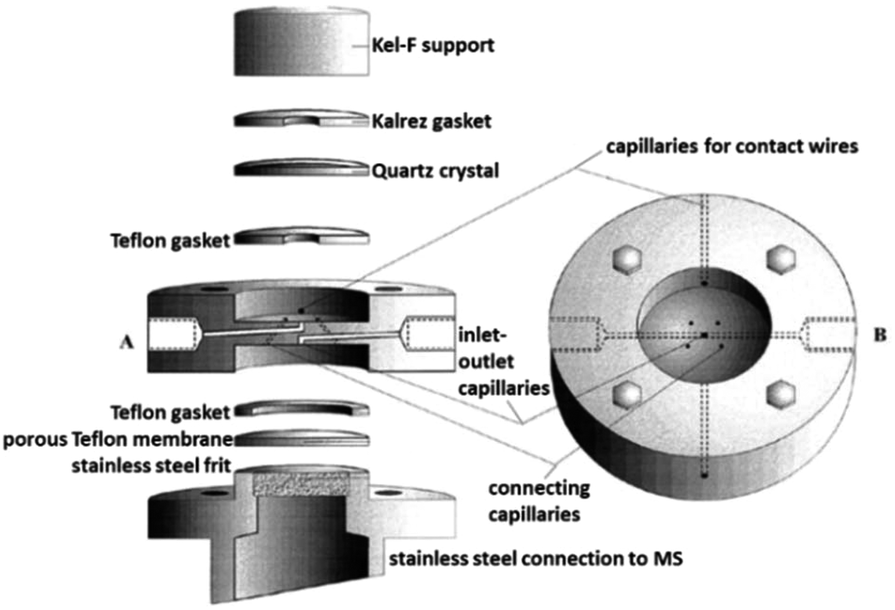 | ||
| Fig. 23 DEMS cell; A: exploded sideview; B: top view. Image republished and modified with permission of Journal of The Electrochemical Society, from the original work by Jusys et al.297 Copyright 1999, permission conveyed through Copyright Clearance Center, Inc. | ||
A simpler one chamber thin-layer cell was reported by Trimarco et al. in 2018.301 Here, they used a previously reported microfabricated Si membrane with supporting SiO2 pillars and an integrated sampling chamber to separate the liquid phase from the gaseous phase.308 The thin-layer cell had a simple one chamber geometry with a WE directly above the membrane chip, allowing for low response times. A CE and RE were positioned in slanted side channels. The membrane chip itself was mounted onto a vacuum system to enable DEMS, shown schematically in Fig. 24. Using this setup, Roy et al. investigated the oxidation evolution reaction catalysed by nanoparticle catalysts.309 Additionally, Winiwarter et al. investigated the mechanisms of partial propene oxidation.310
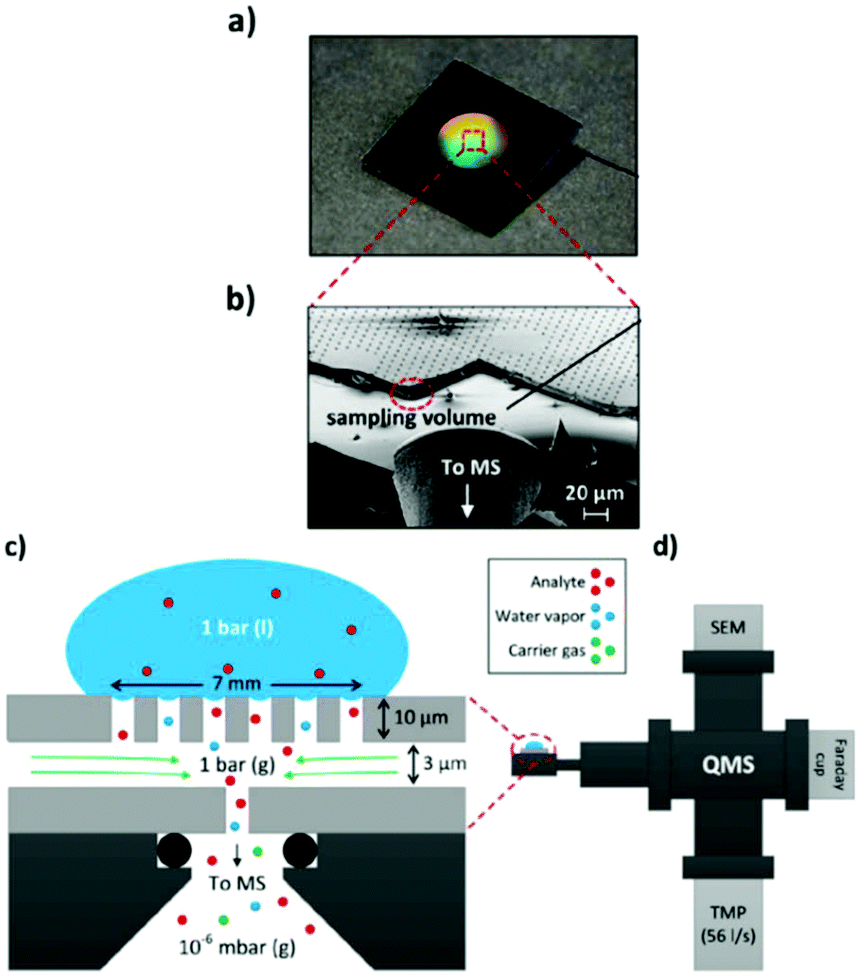 | ||
| Fig. 24 Membrane DEMS setup for thin-layer cell; (a) membrane chip photograph; (b) scanning electron microscope image of the membrane chip; (c) schematic of the membrane chip; (d) schematic of the setup. Image reprinted and modified with permission from Trimarco et al.301 Copyright 2018 American Chemical Society. | ||
5.2.2.2 Microfluidic DEMS. In a similar fashion to the ESI method, a microfluidic approach for DEMS has recently been reported. In 2015, Trimarco et al. reported a “sniffer chip” assembly that uses the previously mentioned microfabricated membrane.308 The microfabrication process is illustrated in Fig. 25. The Si and SiO2 based membrane was incorporated into a microfluidic chip with integrated electrodes a small total internal volume of ca. 31 nL and a low response time about 0.5 s. Trimarco et al. made proof of principle measurements of hydrogen and oxygen evolution reactions in an aqueous solvent. An envisioned application is to use the chip as a dip-in probe that immediately saturates with the gasses in the solution to provide a quick qualitative readout of the sample.
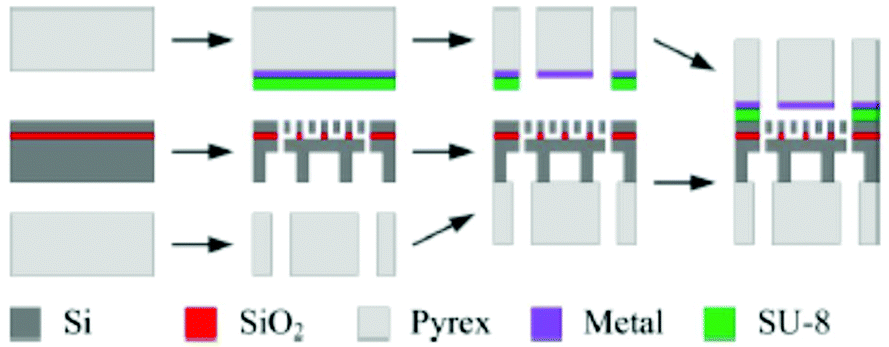 | ||
| Fig. 25 Membrane fabrication scheme. Reprinted from the original work by Trimarco et al.,308 Copyright 2015, with the permission of AIP Publishing. | ||
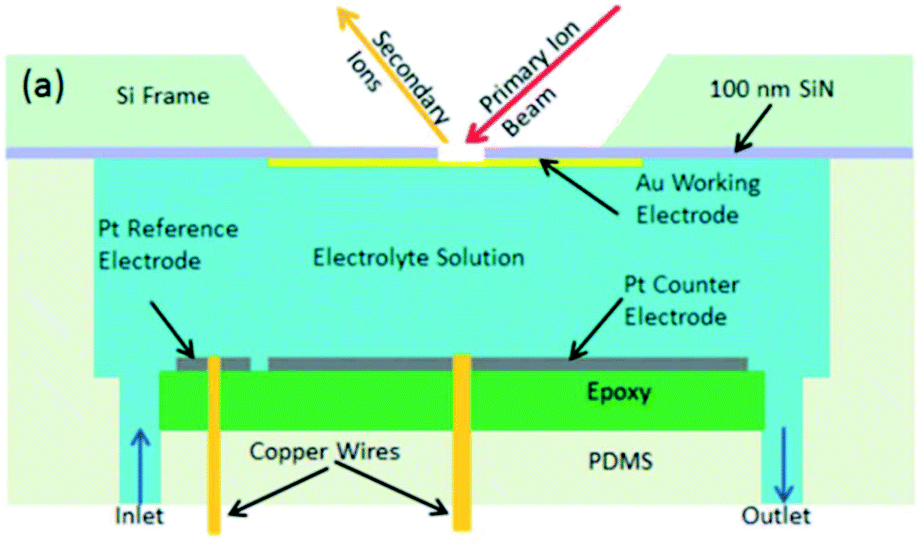 | ||
| Fig. 26 Schematic of an EC-SIMS cell. Reprinted from the original work by Liu et al.,311 Copyright 2014 – Published by The Royal Society of Chemistry. | ||
5.3 Future perspective
We have shown several established and innovative approaches to couple electrochemical devices to mass spectrometers. Table 2 shows the different techniques and semi-quantitative statements about their reported response times, advantages and restrictions.| Ionization method/technique | Response time | Advantages | Drawbacks | Ref. |
|---|---|---|---|---|
| ESI, thin-layer | ∼25 s | Standardized, exchangeable electrode | Long transfer times, special care in gasket handling | 289 |
| ESI, microfluidic | ∼4.5 s | Optimized design, very low analyte amounts | Limited flexibility, difficult fabrication | 287 and 290 |
| ESI, droplet | fast enough to detect intermediates | Very small footprint, simple fabrication | Static experiments, poor reproducibility | 292 |
| ESI, double-barrel capillary | ≤23 ms | Extremely fast, small footprint | Complicated fabrication and operation | 294 and 295 |
| DEMS, thin-layer | 1–2 s | Simple operation, measures formation rate | Long transfer times, restricted to volatile analytes | 297, 301–307, 309 and 310 |
| DEMS, microfluidic | ∼0.5 s | Simple and fast operation, very small footprint | Restricted to volatile analytes, difficult fabrication | 308 |
| SIMS, microfluidic | <100 ms | Very fast and sensitive, can probe electrode/electrolyte interfaces | Small probed area, destroys the WE | 311–313 |
Compared to other spectroelectrochemical methods, however, EC-MS is not being researched to the same degree. As stated previously, the most important part in coupling these techniques is the ionization technique.
The already established microfluidic ESI methods described previously mostly use commercially available nano-ESI needles. A simple way to improve on the methods could lie in the monolithic fabrication of the device. By integrating the needle into the chip design, it could be simpler to setup, avoiding damage to the nano-ESI needle and possible leaks. More importantly, it reduces dead volume. Additionally, the influence of the needle geometry on the ionization success could be investigated more systematically. This has been demonstrated in a relatively straightforward manner by the group of Girault,314,315 using a single carbon paste electrode. To the best of our knowledge it has not been demonstrated with a three-electrode microchip yet.
Additionally, there have been ESI setups where the capillary itself served as an electrochemical reactor in the past.286,316 This approach promises the lowest possible response times and simplest setups and should be investigated further even in present times.
The microfluidic possibilities of using EC-DEMS chips to monitor EC reactions are still only emerging. A spin-off company, SpectroInlets, has created a dedicated system to use their membrane chip technology. It is to be expected that microfluidic DEMS will be a topic of future research.
Lastly, as a general note, there are limitations to the use of ESI and DEMS. While DEMS is strongly restricted to volatile analytes, ESI can struggle with the ionization of certain classes of compounds and ion suppression effects.279 It is therefore also important to investigate other already established ionization methods as a coupling partner to EC. We presented an example that employed SIMS to obtain impressive information about electrode/electrolyte interfaces and could also take short snapshots of intermediates. There are, however, more established and likely also suitable techniques like e.g. atmospheric pressure chemical ionization (APCI), atmospheric pressure photoionization (APPI), atmospheric pressure plasma jet (APPJ) or ICP ionization, the latter having been already mentioned in section 5.2 for methods where species information is not important. These methods mostly use external gasses to nebulize and/or ionize the analytes and thus require additional instrumentation outside the EC cell. They can, however, offer better ionization efficiencies or better versatility than other ionization methods that require less instrumentation because they use different mechanisms to create the ions necessary for MS.
6 Concluding remarks
6.1 IR-SEC
We expect that the advent of QCL lasers will change the future of IR-SEC, solving the inherent problems of low intensity light sources. The QCL lasers will make laser IR spectroscopy more accessible, allowing for faster analysis times. The higher source power will alleviate the need for using the thin-layer configurations and at the same time push the field of ATR IR-SEC towards the use of waveguides. This, together with advancements in nanofabrication will create the possibility of constructing complete IR-SEC lab on chip systems.By making use of IR-transparent conductive waveguides, both the waveguide and the electrode can be combined into one structure. The footprint of the device could be made small enough to be installed as sensing systems in organs on chip, as wearable sensors or in miniaturized high throughput screening platforms.
6.2 Raman-SEC
Unlike IR-SEC, Raman-SEC is already a mature technique. However, especially the field of SERS-SEC can be further developed by combining it with new advancements in the field of nanofabrication. These advancements will possibly lead to developments where SERS enhancement structures can give more reliable and uniform enhancements and at the same time increase the area of the enhancement structures and simultaneously decrease the cost.At the moment Raman-SEC is mostly used for a large number of applications in material sciences, but as of yet does not have a large number of real world applications, some notable exceptions are for instance the work by the group of Brosseau.136–139 When the advancements of SERS-SEC as highlighted above take place, the technique will likely become more attractive for real world applications.
6.3 NMR-SEC
NMR spectroscopy is a very versatile technique that still has much potential to grow and mature. As a consequence, NMR-SEC is also still in its infancy and will benefit from general NMR improvements. There are, however, certain NMR-SEC specific recommendations that can be made.Because of the unique restrictions caused by the interference between NMR and EC systems, dedicated NMR-SEC instruments should be investigated. Also, standardizing or making the often used three-electrode cell type commercially available would be beneficial to make research from different groups both more comparable and reliable.
NMR-SEC can already be used for real-time 1H measurements, but especially real-time 13C measuring methods still need to be improved. Research approaches could be based on sample preparation techniques or the exploitation of NMR-SEC specific effects like magnetoelectrolysis to reduce the time needed for an NMR-SEC experiment.
6.4 EC-MS
Although research towards EC-MS is not being performed to the same degree as the SEC techniques covered in this review, clear paths for future advancements can be discussed. Any improvements on EC-MS regarding the response time between reactions at the electrodes and the ionization of the sample would make the technique more attractive. Directly combining the ionization mechanism, e.g. ESI, with a microfluidic system via new microfabrication techniques could result in monolithic devices with very short response times. Additional ionization techniques that are used for MS but could also be of interest for EC-MS are SIMS, APCI, APPI, APPJ and ICP, as partly already mentioned previously. These other ionization techniques could help to increase the ionization efficiency of certain analytes while also mostly not suffering from the same coupling problems of EC with MS as is the case with ESI. Some of them have their own application restrictions, however, making a careful selection all the more important.6.5 Hyphenation
Hyphenation of the (already hyphenated) SEC techniques, also known as multi-spectroelectrochemistry, is quite common in literature. In 2011 Dunsh wrote a review paper on this very topic.3As the field of SEC advances, so will the number of hyphenation of different techniques. Already in 2011 Dunsch predicted a greater variety in triple multi-spectroelectrochemistry. In order to get more information from a system, we expect that more and more techniques will be combined into multi-spectroelectrochemistry in the future.
Author contributions
Authors J. J. A. Lozeman and P. Führer contributed equally to this review paper. J. J. A. Lozeman was responsible for IR-SEC and Raman-SEC, P. Führer had the responsibility for NMR-SEC and EC-MS. M. Odijk and W. Olthuis supervised and managed the review and edited the final version of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), the Netherlands Organisation for Scientific Research (NWO) Gravitation programme funded by the Ministry of Education, Culture and Science of the government of the Netherlands and the NWO Open Technology Research Programme with project number 15230.The authors would like to thank Esther Tanumihardja and Elsbeth G.B.M. Bossink (BIOS lab-on-a-Chip Group, University of Twente) for their discussions about electrochemistry.
References
- W. Kaim and J. Fiedler, Chem. Soc. Rev., 2009, 38, 3373–3382 RSC.
- T. Kuwana, R. K. Darlington and D. W. Leedy, Anal. Chem., 1964, 36, 2023–2025 CrossRef CAS.
- L. Dunsch, J. Solid State Electrochem., 2011, 15, 1631–1646 CrossRef CAS.
- H. Oberacher, F. Pitterl, R. Erb and S. Plattner, Mass Spectrom. Rev., 2015, 34, 64–92 CrossRef CAS PubMed.
- A. J. Wain and M. A. O'connell, Adv. Phys.: X, 2017, 2, 188–209 CAS.
- Y. Y. J. Tong, Curr. Opin. Electrochem., 2017, 4, 60–68 CrossRef CAS.
- L. León and J. D. Mozo, TrAC, Trends Anal. Chem., 2018, 102, 147–169 CrossRef.
- Y. Zhai, Z. Zhu, S. Zhou, C. Zhu and S. Dong, Nanoscale, 2018, 10, 3089–3111 RSC.
- J. Garoz-Ruiz, J. V. Perales-Rondon, A. Heras and A. Colina, Electroanalysis, 2019, 1254–1278 CrossRef CAS.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. A. Vyvyan, Introduction to Spectroscopy, Cengage Learning, 2008 Search PubMed.
- D. Moss, E. Nabedryk, J. Breton and W. Mantele, Eur. J. Biochem., 1990, 187, 565–572 CrossRef CAS.
- F. T. G. van den Brink, W. Olthuis, A. van den Berg and M. Odijk, TrAC, Trends Anal. Chem., 2015, 70, 40–49 CrossRef CAS.
- P. A. Christensen and S. W. M. Jones, J. Phys. Chem. C, 2014, 118, 29760–29769 CrossRef CAS.
- K. H. K. L. Alwis, M. R. Mucalo and B. Ingham, J. Electrochem. Soc., 2013, 160, 803–812 CrossRef.
- A. L. De Lacey, E. C. Hatchikian, A. Volbeda, M. Frey, J. C. Fontecilla-Camps and V. M. Fernandez, J. Am. Chem. Soc., 1997, 119, 7181–7189 CrossRef CAS.
- R. Guidelli, Bioelectrochemistry of Biomembranes and Biomimetic Membranes, 2016 CAS.
- A. Hartstein, J. R. Kirtley and J. C. Tsang, Phys. Rev. Lett., 1980, 45, 201–204 CrossRef CAS.
- M. Osawa, in Near-Field Optics and Surface Plasmon Polaritons, ed. S. Kawata, Springer Berlin Heidelberg, Berlin, Heidelberg, 2001, pp. 163–187 Search PubMed.
- R. Aroca, Surface-Enhanced Vibrational Spectroscopy, John Wiley & Sons, Ltd, Chichester, UK, 2006 Search PubMed.
- Y. Hu, Á. I. López-Lorente and B. Mizaikoff, ACS Photonics, 2019, 6, 2182–2197 CrossRef CAS.
- K. I. Ataka, T. Yotsuyanagi and M. Osawa, J. Phys. Chem., 1996, 100, 10664–10672 CrossRef CAS.
- M. Osawa, K. Ataka, K. Yoshii and T. Yotsuyanagi, J. Electron Spectrosc. Relat. Phenom., 1993, 64–65, 371–379 CrossRef CAS.
- H. Im, K. C. Bantz, N. C. Lindquist, C. L. Haynes and S. H. Oh, Nano Lett., 2010, 10, 2231–2236 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, Principles of Surface-Enhanced Raman Spectroscopy, Elsevier, 2009 Search PubMed.
- K. Ataka and J. Heberle, Anal. Bioanal. Chem., 2007, 388, 47–54 CrossRef CAS PubMed.
- R. Aroca, Surface-Enhanced Vibrational Spectroscopy, Wiley, 2006 Search PubMed.
- F. Neubrech, C. Huck, K. Weber, A. Pucci and H. Giessen, Chem. Rev., 2017, 117, 5110–5145 CrossRef CAS PubMed.
- C. Di Bari, N. Mano, S. Shleev, M. Pita and A. L. De Lacey, J. Biol. Inorg. Chem., 2017, 22, 1179–1186 CrossRef CAS PubMed.
- Y. Kato and T. Noguchi, Biochemistry, 2014, 53, 4914–4923 CrossRef CAS.
- F. Tutunea and M. D. Ryan, J. Electroanal. Chem., 2012, 670, 16–22 CrossRef CAS.
- F. Tutunea, A. Atifi and M. D. Ryan, J. Electroanal. Chem., 2015, 744, 17–24 CrossRef CAS.
- R. W. Murray, W. R. Heineman and G. W. O'Dom, Anal. Chem., 1967, 39, 1666–1668 CrossRef CAS.
- Z. Wei and M. D. Ryan, Inorg. Chem., 2010, 49, 6948–6954 CrossRef CAS PubMed.
- Y. Dai, D. A. Proshlyakov, J. K. Zak and G. M. Swain, Anal. Chem., 2007, 79, 7526–7533 CrossRef CAS PubMed.
- J. K. Zak, J. E. Butler and G. M. Swain, Anal. Chem., 2001, 73, 908–914 CrossRef CAS PubMed.
- J. Stotter, J. Zak, Z. Behler, Y. Show and G. M. Swain, Anal. Chem., 2002, 74, 5924–5930 CrossRef CAS PubMed.
- K. H. K. L. Alwis, M. R. Mucalo and J. R. Lane, RSC Adv., 2015, 5, 31815–31825 RSC.
- M. J. Shaw, R. L. Henson, S. E. Houk, J. W. Westhoff, M. W. Jones and G. B. Richter-Addo, J. Electroanal. Chem., 2002, 534, 47–53 CrossRef CAS.
- N. Xu, J. Lilly, D. R. Powell and G. B. Richter-Addo, Organometallics, 2012, 31, 827–834 CrossRef CAS.
- M. A. El-Attar, N. Xu, D. Awasabisah, D. R. Powell and G. B. Richter-Addo, Polyhedron, 2012, 40, 105–109 CrossRef CAS.
- D. Awasabisah, N. Xu, K. P. S. Gautam, D. R. Powell, M. J. Shaw and G. B. Richter-Addo, Eur. J. Inorg. Chem., 2016, 2016, 509–518 CrossRef CAS.
- S. Pons, J. Electroanal. Chem., 1983, 150, 495–504 CrossRef CAS.
- D. Aurbach and O. Chusid, J. Electrochem. Soc., 1993, 140, L155–L157 CrossRef CAS.
- M. R. Mucalo and Q. Li, J. Colloid Interface Sci., 2004, 269, 370–380 CrossRef CAS.
- X. Fang, L. Wang, P. K. Shen, G. Cui and C. Bianchini, J. Power Sources, 2010, 195, 1375–1378 CrossRef CAS.
- L. Liu, L. Zeng, L. Wu and X. Jiang, Anal. Chem., 2017, 89, 2724–2730 CrossRef CAS PubMed.
- L. Liu, L. Zeng, L. Wu and X. Jiang, J. Phys. Chem. C, 2015, 119, 3990–3999 CrossRef CAS.
- Y. Y. Yang, J. Ren, H. X. Zhang, Z. Y. Zhou, S. G. Sun and W. Bin Cai, Langmuir, 2013, 29, 1709–1716 CrossRef CAS PubMed.
- L. Wang, H. Meng, P. K. Shen, C. Bianchini, F. Vizza and Z. Wei, Phys. Chem. Chem. Phys., 2011, 13, 2667–2673 RSC.
- Q. Zhang, W. L. Song, A. M. Showkot Hossain, Z. Di Liu, G. J. Hu, Y. P. Tian, J. Y. Wu, B. K. Jin, H. P. Zhou, J. X. Yang and S. Y. Zhang, Dalton Trans., 2011, 40, 3510–3516 RSC.
- W. G. Golden, D. S. Dunn and J. Overend, J. Catal., 1981, 71, 395–404 CrossRef CAS.
- P. W. Faguy and W. N. Richmond, J. Electroanal. Chem., 1996, 410, 109–113 CrossRef.
- P. W. Faguy, W. N. Richmond, R. S. Jackson, S. C. Weibel, G. Ball and J. H. Payer, Appl. Spectrosc., 1998, 52, 557–564 CrossRef CAS.
- M. J. Green, B. J. Barner and R. M. Corn, Rev. Sci. Instrum., 1991, 62, 1426–1430 CrossRef CAS.
- B. J. Barner, M. J. Green, E. I. Sáez and R. M. Corn, Anal. Chem., 1991, 63, 55–60 CrossRef CAS.
- V. Zamlynny, I. Zawisza and J. Lipkowski, Langmuir, 2003, 19, 132–145 CrossRef CAS.
- I. Zawisza, X. Bin and J. Lipkowski, Langmuir, 2007, 23, 5180–5194 CrossRef CAS PubMed.
- N. Garcia-Araez, C. L. Brosseau, P. Rodriguez and J. Lipkowski, Langmuir, 2006, 22, 10365–10371 CrossRef CAS PubMed.
- R. C. Alkire, D. M. Kolb, J. Lipkowski and P. N. Ross, Diffraction and Spectroscopic Methods in Electrochemistry, Wiley, 2009 Search PubMed.
- E. A. Monyoncho, V. Zamlynny, T. K. Woo and E. A. Baranova, Analyst, 2018, 143, 2563–2573 RSC.
- T. Wang, J. Bai, X. Jiang and G. U. Nienhaus, ACS Nano, 2012, 6, 1251–1259 CrossRef CAS PubMed.
- P. Hosseini, G. Wittstock and I. Brand, J. Electroanal. Chem., 2018, 812, 199–206 CrossRef CAS.
- A. J. Bard, Electroanalytical Chemistry: A Series of Advances, Taylor & Francis, 1986 Search PubMed.
- G. Neophytides, L. Quaroni, F. N. Büchi, A. Orfanidi, S. G. Neophytides and T. J. Schmidt, Electrochem. Commun., 2013, 34, 200–203 CrossRef CAS.
- P. Su, V. Prabhakaran, G. E. Johnson and J. Laskin, Anal. Chem., 2018, 90, 10935–10942 CrossRef CAS PubMed.
- M. Villalba, L. P. M. De Leo and E. J. Calvo, ChemElectroChem, 2014, 1, 195–199 CrossRef.
- R. Aroca, D. Ross and C. Domingo, Appl. Spectrosc., 2004, 58, 324A–338A CrossRef CAS PubMed.
- T. Schadle and B. Mizaikoff, Appl. Spectrosc., 2016, 70, 1625–1638 CrossRef PubMed.
- M. Sieger and B. Mizaikoff, Anal. Chem., 2016, 88, 5562–5573 CrossRef CAS PubMed.
- B. K. Purushothaman, M. Pelsozy, P. W. Morrison, V. F. Lvovich and H. B. Martin, J. Appl. Electrochem., 2012, 42, 111–120 CrossRef CAS.
- P. Paengnakorn, P. A. Ash, S. Shaw, K. Danyal, T. Chen, D. R. Dean, L. C. Seefeldt and K. A. Vincent, Chem. Sci., 2017, 8, 1500–1505 RSC.
- A. Hatta, Y. Suzuki, T. Wadayama and W. Suëtaka, Appl. Surf. Sci., 1991, 48–49, 222–226 CrossRef CAS.
- E. Kretschmann, Z. Phys., 1971, 241, 313–324 CrossRef CAS.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- R. Aroca and B. Price, J. Phys. Chem. B, 1997, 101, 6537–6540 CrossRef CAS.
- A. J. Healy, P. A. Ash, O. Lenz and K. A. Vincent, Phys. Chem. Chem. Phys., 2013, 15, 7055–7059 RSC.
- C. Dillard, A. Singh and V. Kalra, J. Phys. Chem. C, 2018, 122, 18195–18203 CrossRef CAS.
- J. Heyes, M. Dunwell and B. Xu, J. Phys. Chem. C, 2016, 120, 17334–17341 CrossRef CAS.
- M. Pfaffeneder-Kmen, I. F. Casas, A. Naghilou, G. Trettenhahn and W. Kautek, Electrochim. Acta, 2017, 255, 160–167 CrossRef CAS.
- A. Viinikanoja, J. Kauppila, P. Damlin, M. Suominen and C. Kvarnström, Phys. Chem. Chem. Phys., 2015, 17, 12115–12123 RSC.
- J. Izquierdo, B. Mizaikoff and C. Kranz, Phys. Status Solidi A, 2016, 213, 2056–2062 CrossRef CAS.
- D. Neubauer, J. Scharpf, A. Pasquarelli, B. Mizaikoff and C. Kranz, Analyst, 2013, 138, 6746–6752 RSC.
- N. Menegazzo, M. Kahn, R. Berghauser, W. Waldhauser and B. Mizaikoff, Analyst, 2011, 136, 1831–1839 RSC.
- S. Sun, S. Hong, S. Chen, G. Lu, H. Dai and X. Xiao, Sci. China, Ser. B: Chem., 1999, 42, 261–267 CrossRef CAS.
- J. T. Li, S. R. Chen, F. S. Ke, G. Z. Wei, L. Huang and S. G. Sun, J. Electroanal. Chem., 2010, 649, 171–176 CrossRef CAS.
- J. T. Li, Q. S. Chen and S. G. Sun, Electrochim. Acta, 2007, 52, 5725–5732 CrossRef CAS.
- H. Gong, S. G. Sun, Y. J. Chen and S. P. Chen, J. Phys. Chem. B, 2004, 108, 11575–11584 CrossRef CAS.
- S.-G. Sun and Z.-Y. Zhou, in In-situ Spectroscopic Studies of Adsorption at the Electrode and Electrocatalysis, ed. S.-G. Sun, P. A. Christensen and A. Wieckowski, Elsevier Science B.V., Amsterdam, 2007, pp. 139–178 Search PubMed.
- C. A. Melendres, B. Beden and G. A. Bowmaker, J. Electroanal. Chem., 1995, 383, 191–193 CrossRef.
- S. M. Rosendahl, F. Borondics, T. E. May, T. M. Pedersen and I. J. Burgess, Anal. Chem., 2011, 83, 3632–3639 CrossRef CAS.
- K. Tu, M. J. Lardner, T. A. Morhart, S. M. Rosendahl, S. Creighton and I. J. Burgess, J. Phys. Chem. C, 2016, 120, 23640–23647 CrossRef CAS.
- S. M. Rosendahl, F. Borondics, T. E. May and I. J. Burgess, Anal. Chem., 2013, 85, 8722–8727 CrossRef CAS.
- M. J. Lardner, K. Tu, B. C. Barlow, S. M. Rosendahl, F. Borondics and I. J. Burgess, J. Electroanal. Chem., 2017, 800, 184–189 CrossRef CAS.
- P. A. Ash, H. A. Reeve, J. Quinson, R. Hidalgo, T. Zhu, I. J. McPherson, M. W. Chung, A. J. Healy, S. Nayak, T. H. Lonsdale, K. Wehbe, C. S. Kelley, M. D. Frogley, G. Cinque and K. A. Vincent, Anal. Chem., 2016, 88, 6666–6671 CrossRef CAS PubMed.
- J. A. Sigrist, E. S. Lins, T. A. Morhart, J. L. Briggs and I. J. Burgess, Appl. Spectrosc., 2019, 73, 1394–1402 CrossRef CAS.
- M. J. Weida and B. Yee, Imaging Manip. Anal. Biomol. Cells Tissues IX, 2011, 7902, 79021C Search PubMed.
- J. M. Ramirez, V. Vakarin, C. Gilles, J. Frigerio, A. Ballabio, P. Chaisakul, X. Le Roux, C. Alonso-Ramos, G. Maisons, L. Vivien, M. Carras, G. Isella and D. Marris-Morini, Opt. Lett., 2017, 42, 105 CrossRef CAS PubMed.
- P. Wägli, Y. C. Chang, A. Homsy, L. Hvozdara, H. P. Herzig and N. F. De Rooij, Anal. Chem., 2013, 85, 7558–7565 CrossRef PubMed.
- P. G.-Q. Lo, A. A. Bettiol, A. E.-J. Lim, K.-W. Ang and U. Younis, IEEE Photonics Technol. Lett., 2016, 28, 2447–2450 Search PubMed.
- C. B. E. Gawith, A. Z. Khokhar, C. Littlejohns, G. S. Murugan, J. S. Penades, V. Mittal, M. Nedeljkovic, J. S. Wilkinson, G. Z. Mashanovich and L. G. Carpenter, Opt. Express, 2017, 25, 27431 CrossRef PubMed.
- N. Singh, A. Casas-Bedoya, D. D. Hudson, A. Read, E. Mägi and B. J. Eggleton, Opt. Lett., 2016, 41, 5776 CrossRef CAS.
- V. Mittal, J. S. Wilkinson and G. S. Murugan, High-contrast GeTe4 waveguides for mid-infrared biomedical sensing applications, ed. J. E. Broquin and G. Nunzi Conti, International Society for Optics and Photonics, 2014, p. 89881A Search PubMed.
- Z. Cheng, X. Chen, C. Y. Wong, K. Xu and H. K. Tsang, IEEE Photonics J., 2012, 4, 1510–1519 Search PubMed.
- Y. C. Chang, P. Wägli, V. Paeder, A. Homsy, L. Hvozdara, P. Van Der Wal, J. Di Francesco, N. F. De Rooij and H. Peter Herzig, Lab Chip, 2012, 12, 3020–3023 RSC.
- M. Malmström, M. Karlsson, P. Forsberg, Y. Cai, F. Nikolajeff and F. Laurell, Opt. Mater. Express, 2016, 6, 1286 CrossRef.
- A. Osman, A. Z. Khokhar, M. Nedeljkovic, Y. Wu, G. Z. Mashanovich, K. Debnath, Z. Qu and J. Soler Penades, Opt. Lett., 2018, 43, 5997 CrossRef CAS.
- Y. Tzeng, Diamond Films Technol., 1991, 1, 31 CAS.
- M. Odijk and A. van den Berg, Annu. Rev. Anal. Chem., 2018, 11, 421–440 CrossRef CAS PubMed.
- A. Susarrey-arce, R. M. Tiggelaar, M. Morassutto, J. Geerlings and R. G. P. Sanders, Sens. Actuators, B, 2015, 220, 13–21 CrossRef CAS.
- R. Hersig-Marx, K. T. Queeney, R. J. Jackman, M. A. Schmidt and K. F. Jensen, Anal. Chem., 2004, 76, 6476–6483 CrossRef PubMed.
- E. Karabudak, B. L. Mojet, S. Schlautmann, G. Mul and H. J. G. E. Gardeniers, Anal. Chem., 2012, 84, 3132–3137 CrossRef CAS PubMed.
- B. A. Rizkin, F. G. Popovic and R. L. Hartman, J. Vac. Sci. Technol., A, 2019, 37, 050801 CrossRef.
- P. Führer, J. J. A. Lozeman, H. L. de Boer, J. G. Bomer, W. Olthuis and M. Odijk, 22nd Int. Conf. Miniaturized Syst. Chem. Life Sci. μTAS, 2018, vol. 2018, pp. 2127–2130.
- E. Gross, Nano Res., 2019, 12, 2200–2210 CrossRef.
- A. Dazzi and C. B. Prater, Chem. Rev., 2017, 117, 5146–5173 CrossRef CAS PubMed.
- Y. H. Lu, J. M. Larson, A. Baskin, X. Zhao, P. D. Ashby, D. Prendergast, H. A. Bechtel, R. Kostecki and M. Salmeron, Nano Lett., 2019, 19, 5388–5393 CrossRef CAS PubMed.
- F. Huth, A. Govyadinov, S. Amarie, W. Nuansing, F. Keilmann and R. Hillenbrand, Nano Lett., 2012, 12, 3973–3978 CrossRef CAS PubMed.
- R. Adato, S. Aksu and H. Altug, Biochem. Pharmacol., 2015, 18, 436–446 CAS.
- T. Schmid and P. Dariz, Heritage, 2019, 2, 1662–1683 CrossRef.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem., 1977, 84, 1 CrossRef CAS.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- M. Moskovits, Phys. Chem. Chem. Phys., 2013, 15, 5301–5311 RSC.
- M. Moskovits, J. Raman Spectrosc., 2005, 36, 485–496 CrossRef CAS.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef.
- J. Kim, Y. Jang, N. J. Kim, H. Kim, G. C. Yi, Y. Shin, M. H. Kim and S. Yoon, Front. Chem., 2019, 7, 1–7 CrossRef PubMed.
- M. A. Montiel, F. J. Vidal-Iglesias, V. Montiel and J. Solla-Gullón, Curr. Opin. Electrochem., 2017, 1, 34–39 CrossRef CAS.
- A. Otto, J. Raman Spectrosc., 1991, 22, 743–752 CrossRef CAS.
- M. F. dos Santos, V. Katic, P. L. dos Santos, B. M. Pires, A. L. B. Formiga and J. A. Bonacin, Anal. Chem., 2019, 91, 10386–10389 CrossRef CAS PubMed.
- D. Martín-Yerga, A. Pérez-Junquera, D. Hernández-Santos and P. Fanjul-Bolado, Electroanalysis, 2018, 30, 1095–1099 CrossRef.
- O. J. R. Clarke, G. J. H. St Marie and C. L. Brosseau, J. Electrochem. Soc., 2017, 164, B3091–B3095 CrossRef CAS.
- C. N. Hernández, D. Martín-Yerga, M. B. González-García, D. Hernández-Santos and P. Fanjul-Bolado, Talanta, 2018, 178, 85–88 CrossRef PubMed.
- D. Martín-Yerga, A. Pérez-Junquera, M. B. González-García, J. V. Perales-Rondon, A. Heras, A. Colina, D. Hernández-Santos and P. Fanjul-Bolado, Electrochim. Acta, 2018, 264, 183–190 CrossRef.
- T. P. Lynk, O. J. R. Clarke, N. Kesavan and C. L. Brosseau, Sens. Actuators, B, 2018, 257, 270–277 CrossRef CAS.
- A. M. Robinson, S. G. Harroun, J. Bergman and C. L. Brosseau, Anal. Chem., 2012, 84, 1760–1764 CrossRef CAS PubMed.
- R. A. Karaballi, A. Nel, S. Krishnan, J. Blackburn and C. L. Brosseau, Phys. Chem. Chem. Phys., 2015, 17, 21356–21363 RSC.
- L. Zhao, J. Blackburn and C. L. Brosseau, Anal. Chem., 2015, 87, 441–447 CrossRef CAS PubMed.
- S. D. Bindesri, D. S. Alhatab and C. L. Brosseau, Analyst, 2018, 143, 4128–4135 RSC.
- G. Niaura, A. K. Gaigalas and V. L. Vilker, J. Raman Spectrosc., 1997, 28, 1009–1011 CrossRef CAS.
- R. Mažeikiená, G. Niaura and A. Malinauskas, Chemija, 2018, 29, 81–88 Search PubMed.
- R. Mažeikiene, G. Niaura and A. Malinauskas, Synth. Met., 2010, 160, 1060–1064 CrossRef.
- R. Mažeikienė, G. Niaura and A. Malinauskas, J. Solid State Electrochem., 2019, 23, 1631–1640 CrossRef.
- R. Mažeikienė, G. Niaura and A. Malinauskas, Synth. Met., 2019, 248, 35–44 CrossRef.
- R. Mažeikienė, G. Niaura and A. Malinauskas, Synth. Met., 2018, 243, 97–106 CrossRef.
- R. Mažeikienė, G. Niaura and A. Malinauskas, Spectrochim. Acta, Part A, 2019, 221, 117147 CrossRef PubMed.
- R. Mažeikiene, G. Niaura, O. Eicher-Lorka and A. Malinauskas, J. Colloid Interface Sci., 2011, 357, 189–197 CrossRef PubMed.
- R. Mažeikien, G. Niaura and A. Malinauskas, J. Electroanal. Chem., 2011, 660, 140–146 CrossRef.
- R. Mažeikiene, G. Niaura and A. Malinauskas, Chemija, 2017, 28, 28–32 Search PubMed.
- R. Mažeikienė, G. Niaura and A. Malinauskas, J. Solid State Electrochem., 2019, 2307–2316 CrossRef.
- R. Mažeikienė, G. Niaura and A. Malinauskas, Spectrochim. Acta, Part A, 2017, 181, 200–207 CrossRef PubMed.
- R. Mažeikiene, G. Niaura and A. Malinauskas, J. Electroanal. Chem., 2014, 719, 60–71 CrossRef.
- W. Kiefer and H. J. Bernstein, Appl. Spectrosc., 1971, 25, 609–613 CrossRef CAS.
- H. Yamada, T. Amamiya and H. Tsubomura, Chem. Phys. Lett., 1978, 56, 591–594 CrossRef CAS.
- P. Hildebrandt, K. A. Macor and R. S. Czernuszewicz, J. Raman Spectrosc., 1988, 19, 65–69 CrossRef CAS.
- A. J. McQuillant and R. E. Hester, J. Raman Spectrosc., 1984, 15, 15–19 CrossRef.
- J. E. Pemberton and R. P. Buck, Appl. Spectrosc., 1981, 35, 571–576 CrossRef CAS.
- J. E. Pemberton and R. P. Buck, J. Electroanal. Chem. Interfacial Electrochem., 1982, 136, 201–208 CrossRef CAS.
- L. Kavan, P. Janda, M. Krause, F. Ziegs and L. Dunsch, Anal. Chem., 2009, 81, 2017–2021 CrossRef CAS PubMed.
- L. Pospíil, M. Gál, M. Hromadová, J. Bulíková, V. Kolivoka, J. Cvaka, K. Nováková, L. Kavan, M. Zukalová and L. Dunsch, Phys. Chem. Chem. Phys., 2010, 12, 14095–14101 RSC.
- P. Damlin, C. Kvarnström, A. Petr, P. Ek, L. Dunsch and A. Ivaska, J. Solid State Electrochem., 2002, 6, 291–301 CrossRef CAS.
- A. Viinikanoja, J. Kauppila, P. Damlin, M. Suominen and C. Kvarnström, Phys. Chem. Chem. Phys., 2015, 17, 12115–12123 RSC.
- M. L. McGlashen, M. E. Blackwood and T. G. Spiro, J. Am. Chem. Soc., 1993, 115, 2074–2075 CrossRef CAS.
- D. Ibañez, D. Plana, A. Heras, D. J. Fermín and A. Colina, Electrochem. Commun., 2015, 54, 14–17 CrossRef.
- F. Bonhomme, J. C. Lassègues and L. Servant, J. Electrochem. Soc., 2001, 148, 450–458 CrossRef.
- L. Kavan and L. Dunsch, ChemPhysChem, 2003, 4, 944–950 CrossRef PubMed.
- M. Kalbáč, L. Kavan, S. Gorantla, T. Gemming and L. Dunsch, Chem. – Eur. J., 2010, 16, 11753–11759 CrossRef PubMed.
- M. Kalbac, L. Kavan, M. Zukalová and L. Dunsch, J. Phys. Chem. C, 2007, 111, 1079–1085 CrossRef CAS.
- M. Kalbac, Y. P. Hsieh, H. Farhat, L. Kavan, M. Hofmann, J. Kong and M. S. Dresselhaus, Nano Lett., 2010, 10, 4619–4626 CrossRef CAS PubMed.
- M. Kalbáč, V. Vales, L. Kavan and L. Dunsch, Nanotechnology, 2014, 25, 485706 CrossRef PubMed.
- M. Kalbac, L. Kavan and L. Dunsch, Diamond Relat. Mater., 2009, 18, 972–974 CrossRef CAS.
- L. Kavan, P. Rapta and L. Dunsch, Chem. Phys. Lett., 2000, 328, 363–368 CrossRef CAS.
- J. Tarábek, L. Kavan, L. Dunsch and M. Kalbac, J. Phys. Chem. C, 2008, 112, 13856–13861 CrossRef.
- L. Kavan, O. Frank, A. A. Green, M. C. Hersam, J. Koltai, V. Zólyomi, J. Kürti and L. Dunsch, J. Phys. Chem. C, 2008, 112, 14179–14187 CrossRef CAS.
- L. Kavan, L. Dunsch and H. Kataura, Chem. Phys. Lett., 2002, 361, 79–85 CrossRef CAS.
- M. Kalbáč, L. Kavan and L. Dunsch, Anal. Chem., 2007, 79, 9074–9081 CrossRef PubMed.
- M. Kalbáč, L. Kavan, M. Zukalová and L. Dunsch, Adv. Funct. Mater., 2005, 15, 418–426 CrossRef.
- C. S. L. Koh, H. K. Lee, G. C. Phan-Quang, X. Han, M. R. Lee, Z. Yang and X. Y. Ling, Angew. Chem., Int. Ed., 2017, 56, 8813–8817 CrossRef CAS PubMed.
- L. Zhang, V. Liao and Z. Yu, Carbon, 2010, 48, 2582–2589 CrossRef CAS.
- D. Ibañez, E. C. Romero, A. Heras and A. Colina, Electrochim. Acta, 2014, 129, 171–176 CrossRef.
- M. Tominaga, N. Watanabe and Y. Yatsugi, J. Electroanal. Chem., 2017, 800, 156–161 CrossRef CAS.
- A. Colina, V. Ruiz, A. Heras, E. Ochoteco, E. Kauppinen and J. López-Palacios, Electrochim. Acta, 2011, 56, 1294–1299 CrossRef CAS.
- R. V. Salvatierra, L. G. Moura, M. M. Oliveira, M. A. Pimenta and A. J. G. Zarbin, J. Raman Spectrosc., 2012, 43, 1094–1100 CrossRef CAS.
- M. Kalbac, H. Farhat, J. Kong, P. Janda, L. Kavan and M. S. Dresselhaus, Nano Lett., 2011, 11, 1957–1963 CrossRef CAS PubMed.
- O. Frank, M. S. Dresselhaus and M. Kalbac, Acc. Chem. Res., 2015, 48, 111–118 CrossRef CAS PubMed.
- J. Binder, J. M. Urban, R. Stepniewski, W. Strupinski and A. Wysmolek, Nanotechnology, 2016, 27, 045704 CrossRef CAS PubMed.
- M. Bousa, O. Frank and L. Kavan, Electroanalysis, 2014, 26, 57–61 CrossRef CAS.
- M. Bouša, O. Frank, I. Jirka and L. Kavan, Phys. Status Solidi B, 2013, 250, 2662–2667 CrossRef.
- S. Gupta, S. B. Carrizosa, J. Jasinski and N. Dimakis, AIP Adv., 2018, 8, 065225 CrossRef.
- W. T. E. van den Beld, M. Odijk, R. H. J. Vervuurt, J. W. Weber, A. A. Bol, A. van den Berg and J. C. T. Eijkel, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- R. Mažeikienė, G. Niaura and A. Malinauskas, J. Electroanal. Chem., 2018, 808, 228–235 CrossRef.
- O. L. Gribkova, O. D. Iakobson, A. A. Nekrasov, V. A. Cabanova, V. A. Tverskoy, A. R. Tameev and A. V. Vannikov, Electrochim. Acta, 2016, 222, 409–420 CrossRef CAS.
- B. Zanfrognini, A. Colina, A. Heras, C. Zanardi, R. Seeber and J. López-Palacios, Polym. Degrad. Stab., 2011, 96, 2112–2119 CrossRef CAS.
- A. Blacha-Grzechnik, K. Piwowar, P. Zassowski, R. Motyka and J. Zak, Electrochim. Acta, 2017, 245, 902–911 CrossRef CAS.
- S. Bilal, A. A. Shah and R. Holze, Vib. Spectrosc., 2010, 53, 279–284 CrossRef CAS.
- S. Shreepathi and R. Holze, Langmuir, 2006, 22, 5196–5204 CrossRef CAS PubMed.
- A. A. Nekrasov, O. L. Gribkova, O. D. Iakobson, I. N. Ardabievskii, V. F. Ivanov and A. V. Vannikov, Chem. Pap., 2017, 71, 449–458 CrossRef CAS.
- Z. Morávková and E. Dmitrieva, J. Raman Spectrosc., 2017, 48, 1229–1234 CrossRef.
- Z. Morávková and E. Dmitrieva, ChemistrySelect, 2019, 4, 8847–8854 CrossRef.
- M. Grzeszczuk, A. Kepas, C. Kvarnstrom and A. Ivaska, Synth. Met., 2010, 160, 636–642 CrossRef CAS.
- K. Crowley and J. Cassidy, J. Electroanal. Chem., 2003, 547, 75–82 CrossRef CAS.
- L. M. C. Ferreira, D. Grasseschi, M. S. F. Santos, P. R. Martins, I. G. R. Gutz, A. M. C. Ferreira, K. Araki, H. E. Toma and L. Angnes, Langmuir, 2015, 31, 4351–4360 CrossRef CAS PubMed.
- A. J. Wilson and K. A. Willets, Analyst, 2016, 141, 5144–5151 RSC.
- C. Xu, H. Geng, R. Bennett, D. A. Clayton and S. Pan, J. Phys. Chem. C, 2013, 117, 1849–1856 CrossRef CAS.
- P. Song, X. Guo, Y. Pan, Y. Wen, Z. Zhang and H. Yang, J. Electroanal. Chem., 2013, 688, 384–391 CrossRef CAS.
- T. Shegai, A. Vaskevich, I. Rubinstein and G. Haran, J. Am. Chem. Soc., 2009, 131, 14392–14398 CrossRef PubMed.
- T. Itoh and R. L. McCreery, J. Am. Chem. Soc., 2002, 124, 10894–10902 CrossRef CAS PubMed.
- W. F. Paxton, S. L. Kleinman, A. N. Basuray, J. F. Stoddart and R. P. Van Duyne, J. Phys. Chem. Lett., 2011, 2, 1145–1149 CrossRef CAS PubMed.
- M. B. Pomfret, J. J. Pietron and J. C. Owrutsky, Langmuir, 2010, 26, 6809–6817 CrossRef CAS PubMed.
- R. A. Karaballi, S. Merchant, S. R. Power and C. L. Brosseau, Phys. Chem. Chem. Phys., 2018, 20, 4513–4526 RSC.
- K. E. R. McLeod, T. P. Lynk, C. S. Sit and C. L. Brosseau, Anal. Methods, 2019, 11, 924–929 RSC.
- D. Ibañez, A. Santidrian, A. Heras, M. Kalbáč and A. Colina, J. Phys. Chem. C, 2015, 119, 8191–8198 CrossRef.
- B. H. C. Greene, D. S. Alhatab, C. C. Pye and C. L. Brosseau, J. Phys. Chem. C, 2017, 121, 8084–8090 CrossRef.
- T. Yuan, L. Le Thi Ngoc, J. Van Nieuwkasteele, M. Odijk, A. van Den Berg, H. Permentier, R. Bischoff and E. T. Carlen, Anal. Chem., 2015, 87, 2588–2592 CrossRef CAS.
- H. Le-The, J. J. A. Lozeman, M. Lafuente, P. Muñoz, J. G. Bomer, H. Duy-Tong, E. Berenschot, A. van den Berg, N. R. Tas, M. Odijk and J. C. T. Eijkel, Nanoscale, 2019, 11, 12152–12160 RSC.
- H. Le-The, R. M. Tiggelaar, E. Berenschot, A. van den Berg, N. Tas and J. C. T. Eijkel, ACS Nano, 2019, 13, 6782–6789 CrossRef CAS.
- J. V. Perales-Rondon, S. Hernandez, A. Heras and A. Colina, Appl. Surf. Sci., 2019, 473, 366–372 CrossRef CAS.
- J. V. Perales-Rondon, S. Hernandez, D. Martin-Yerga, P. Fanjul-Bolado, A. Heras and A. Colina, Electrochim. Acta, 2018, 282, 377–383 CrossRef CAS.
- A. J. Bard and M. Stratmann, Encyclopedia of electrochemistry, 2007, ISBN: 9783527610426, DOI:10.1002/9783527610426.
- C. Zong, C. J. Chen, M. Zhang, D. Y. Wu and B. Ren, J. Am. Chem. Soc., 2015, 137, 11768–11774 CrossRef CAS PubMed.
- C. Shi, W. Zhang, R. L. Birke and J. R. Lombardi, J. Electroanal. Chem., 1997, 423, 67–81 CrossRef CAS.
- D. H. Murgida and P. Hildebrandt, J. Am. Chem. Soc., 2001, 123, 4062–4068 CrossRef CAS PubMed.
- D. H. Murgida and P. Hildebrandt, Angew. Chem., Int. Ed., 2001, 40, 728–731 CrossRef CAS PubMed.
- C. Shi, W. Zhang, J. R. Lombardi and R. L. Birke, J. Phys. Chem., 1992, 96, 10093–10096 CrossRef CAS.
- W. Zhang, A. Vivoni, J. R. Lombardi and R. L. Birke, J. Phys. Chem., 1995, 99, 12846–12857 CrossRef CAS.
- C. Shi, W. Zhang, R. L. Birke and J. R. Lombardi, J. Phys. Chem., 1990, 94, 4766–4769 CrossRef CAS.
- J. Wessel, J. Opt. Soc. Am. B, 1985, 2, 1538–1541 CrossRef CAS.
- R. M. Stöckle, Y. D. Suh, V. Deckert and R. Zenobi, Chem. Phys. Lett., 2000, 318, 131–136 CrossRef.
- M. S. Anderson, Appl. Phys. Lett., 2000, 76, 3130–3132 CrossRef CAS.
- N. Hayazawa, Y. Inouye, Z. Sekkat and S. Kawata, Opt. Commun., 2000, 183, 333–336 CrossRef CAS.
- B. Pettinger, G. Picardi, R. Schuster and G. Ertl, Electrochemistry, 2000, 68, 942–949 CrossRef CAS.
- F. Shao and R. Zenobi, Anal. Bioanal. Chem., 2019, 411, 37–61 CrossRef CAS PubMed.
- D. Kurouski, M. Mattei and R. P. Van Duyne, Nano Lett., 2015, 15, 7956–7962 CrossRef CAS PubMed.
- Z. C. Zeng, S. C. Huang, D. Y. Wu, L. Y. Meng, M. H. Li, T. X. Huang, J. H. Zhong, X. Wang, Z. L. Yang and B. Ren, J. Am. Chem. Soc., 2015, 137, 11928–11931 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- J. F. Li, Y. J. Zhang, A. V. Rudnev, J. R. Anema, S. B. Li, W. J. Hong, P. Rajapandiyan, J. Lipkowski, T. Wandlowski and Z. Q. Tian, J. Am. Chem. Soc., 2015, 137, 2400–2408 CrossRef CAS PubMed.
- L. A. Colnago, F. D. Andrade, A. A. Souza, R. B. V. Azeredo, A. A. Lima, L. M. Cerioni, T. M. Osán and D. J. Pusiol, Chem. Eng. Technol., 2014, 37, 191–203 CrossRef CAS.
- L. M. Silva Nunes, T. B. Moraes, L. L. Barbosa, L. H. Mazo and L. A. Colnago, Anal. Chim. Acta, 2014, 850, 1–5 CrossRef PubMed.
- T. Kobayashi, P. K. Babu, J. H. Chung, E. Oldfield and A. Wieckowski, J. Phys. Chem. C, 2007, 111, 7078–7083 CrossRef CAS.
- P. K. Babu, J. H. Chung, E. Oldfield and A. Wieckowski, Electrochim. Acta, 2008, 53, 6672–6679 CrossRef CAS.
- P. K. Babu, H. S. Kim, J. H. Chung, E. Oldfield and A. Wieckowski, J. Phys. Chem. B, 2004, 108, 20228–20232 CrossRef CAS.
- J. B. Day, P. A. Vuissoz, E. Oldfield, A. Wieckowski and J. P. Ansermet, J. Am. Chem. Soc., 1996, 118, 13046–13050 CrossRef CAS.
- P. K. Babu, H. S. Kim, E. Oldfield and A. Wieckowski, J. Phys. Chem. B, 2003, 107, 7595–7600 CrossRef CAS.
- Y. Y. Tong, S. K. Hee, P. K. Babu, P. Waszczuk, A. Wieckowski and E. Oldfield, J. Am. Chem. Soc., 2002, 124, 468–473 CrossRef CAS PubMed.
- Y. Y. Tong, C. Rice, A. Wieckowski and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 1123–1129 CrossRef CAS.
- Y. Y. Tong, C. Rice, A. Wieckowski and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 11921–11924 CrossRef CAS.
- Y. Tong, C. Rice, N. Godbout, A. Wieckowski and E. Oldfield, J. Am. Chem. Soc., 1999, 121, 2996–3003 CrossRef CAS.
- C. Rice, Y. Tong, E. Oldfield and A. Wieckowski, Electrochim. Acta, 1998, 43, 2825–2830 CrossRef CAS.
- H. Yano, J. Inukai, H. Uchida, M. Watanabe, P. K. Babu, T. Kobayashi, J. H. Chung, E. Oldfield and A. Wieckowski, Phys. Chem. Chem. Phys., 2006, 8, 4932–4939 RSC.
- Y. Y. Tong, A. Wieckowski and E. Oldfield, J. Phys. Chem. B, 2002, 106, 2434–2446 CrossRef CAS.
- P. A. Vuissoz, J. P. H. Ansermet and A. Wieckowski, Phys. Rev. Lett., 1999, 83, 2457–2460 CrossRef CAS.
- P. J. Slezak and A. Wieckowski, J. Electroanal. Chem., 1992, 339, 401–410 CrossRef CAS.
- S. Klod, F. Ziegs and L. Dunsch, Anal. Chem., 2009, 81, 10262–10267 CrossRef CAS PubMed.
- J. Z. Hu, N. R. Jaegers, M. Y. Hu and K. T. Mueller, J. Phys.: Condens. Matter, 2018, 30, 463001 CrossRef PubMed.
- R. D. Webster, Anal. Chem., 2004, 76, 1603–1610 CrossRef CAS PubMed.
- X. Zhang and J. W. Zwanziger, J. Magn. Reson., 2011, 208, 136–147 CrossRef CAS PubMed.
- S. Klod and L. Dunsch, Magn. Reson. Chem., 2011, 49, 725–729 CrossRef CAS PubMed.
- P. Rapta, K. Haubner, P. Machata, V. Lukeš, M. Rosenkranz, S. Schiemenz, S. Klod, H. Kivelä, C. Kvarnström, H. Hartmann and L. Dunsch, Electrochim. Acta, 2013, 110, 670–680 CrossRef CAS.
- M. Zalibera, P. Machata, T. T. Clikeman, M. Rosenkranz, S. H. Strauss, O. V. Boltalina and A. A. Popov, Analyst, 2015, 140, 7209–7216 RSC.
- L. Huang, E. G. Sorte, S. G. Sun and Y. Y. J. Tong, Chem. Commun., 2015, 51, 8086–8088 RSC.
- U. Bussy, P. Giraudeau, V. Silvestre, T. Jaunet-Lahary, V. Ferchaud-Roucher, M. Krempf, S. Akoka, I. Tea and M. Boujtita, Anal. Bioanal. Chem., 2013, 405, 5817–5824 CrossRef CAS PubMed.
- R. Boisseau, U. Bussy, P. Giraudeau and M. Boujtita, Anal. Chem., 2015, 87, 372–375 CrossRef CAS PubMed.
- B. Ferreira Gomes, P. Ferreira da Silva, C. M. Silva Lobo, M. da Silva Santos and L. A. Colnago, Anal. Chim. Acta, 2017, 983, 91–95 CrossRef PubMed.
- E. J. Kelly, J. Electrochem. Soc., 1977, 124, 987–994 CrossRef CAS.
- B. Ferreira Gomes, L. M. Silva Nunes, C. M. Silva Lobo, L. F. Cabeça and L. A. Colnago, Anal. Chem., 2014, 86, 9391–9393 CrossRef PubMed.
- L. M. A. Monzon and J. M. D. Coey, Electrochem. Commun., 2014, 42, 38–41 CrossRef CAS.
- D. W. Mincey, M. J. Popovich, P. J. Faustino, M. M. Hurst and J. A. Caruso, Anal. Chem., 1990, 62, 1197–1200 CrossRef CAS.
- S. H. Cao, Z. R. Ni, L. Huang, H. J. Sun, B. Tang, L. J. Lin, Y. Q. Huang, Z. Y. Zhou, S. G. Sun and Z. Chen, Anal. Chem., 2017, 89, 3810–3813 CrossRef CAS PubMed.
- E. G. Sorte and Y. J. Tong, J. Electroanal. Chem., 2016, 769, 1–4 CrossRef CAS.
- E. G. Sorte, S. Jilani and Y. Y. J. Tong, Electrocatalysis, 2017, 8, 95–102 CrossRef CAS.
- X. P. Zhang, W. L. Jiang, S. H. Cao, H. J. Sun, X. Q. You, S. H. Cai, J. L. Wang, C. Sen Zhao, X. Wang, Z. Chen and S. G. Sun, Electrochim. Acta, 2018, 273, 300–306 CrossRef CAS.
- J. Wang, X. Cao, L. Fang, X. You, K. Wong, S. Cao, C. Xiao, S. Cai, Y. Huang, X. Zhang and Z. Chen, Int. J. Hydrogen Energy, 2019, 44, 16411–16423 CrossRef CAS.
- J. Wang, X. You, C. Xiao, X. Zhang, S. Cai, W. Jiang, S. Guo, S. Cao and Z. Chen, Appl. Catal., B, 2019, 259, 118060 CrossRef CAS.
- S. H. Cao, S. Liu, H. J. Sun, L. Huang, Z. R. Ni, W. L. Jiang, M. Zhan, Z. Y. Zhou, S. G. Sun and Z. Chen, Anal. Chem., 2019, 91, 1686–1691 CrossRef CAS PubMed.
- P. Ferreira da Silva, B. Ferreira Gomes, C. M. Silva Lobo, L. H. K. Queiroz Júnior, E. Danieli, M. Carmo, B. Blümich and L. A. Colnago, Microchem. J., 2019, 146, 658–663 CrossRef CAS.
- J. B. Richter, C. Eßbach, I. Senkovska, S. Kaskel and E. Brunner, Chem. Commun., 2019, 55, 6042–6045 RSC.
- H. Wang, T. K. J. Köster, N. M. Trease, J. Ségalini, P. L. Taberna, P. Simon, Y. Gogotsi and C. P. Grey, J. Am. Chem. Soc., 2011, 133, 19270–19273 CrossRef CAS PubMed.
- B. Ferreira Gomes, C. M. Silva Lobo and L. A. Colnago, Appl. Sci., 2019, 9, 498 CrossRef.
- A. P. Bruins, TrAC, Trends Anal. Chem., 2015, 70, 14–19 CrossRef CAS.
- C. Dass, Fundamentals of Contemporary Mass Spectrometry, Wiley, 2007 Search PubMed.
- W. Plieth, G. S. Wilson and C. Gutiérrez De La Fe, Pure Appl. Chem., 1998, 70, 1395–1414 CAS.
- O. Kasian, S. Geiger, K. J. J. Mayrhofer and S. Cherevko, Chem. Rec., 2019, 19, 2130–2142 CrossRef CAS PubMed.
- S. O. Klemm, A. A. Topalov, C. A. Laska and K. J. J. Mayrhofer, Electrochem. Commun., 2011, 13, 1533–1535 CrossRef CAS.
- S. O. Klemm, A. Karschin, A. K. Schuppert, A. A. Topalov, I. Katsounaros and K. J. J. Mayrhofer, J. Electroanal. Chem., 2013, 693, 127 CrossRef CAS.
- E. M. Yuill and L. A. Baker, ChemElectroChem, 2017, 4, 806–821 CrossRef CAS.
- X. Xu, W. Lu and R. B. Cole, Anal. Chem., 1996, 2700, 4244–4253 CrossRef PubMed.
- F. T. G. van den Brink, L. Büter, M. Odijk, W. Olthuis, U. Karst and A. van den Berg, Anal. Chem., 2015, 87, 1527–1535 CrossRef CAS PubMed.
- H. Faber, M. Vogel and U. Karst, Anal. Chim. Acta, 2014, 834, 9–21 CrossRef CAS PubMed.
- R. Zabel and G. Weber, Anal. Bioanal. Chem., 2016, 408, 1237–1247 CrossRef CAS PubMed.
- M. Odijk, W. Olthuis, A. van den Berg, L. Qiao and H. Girault, Anal. Chem., 2012, 84, 9176–9183 CrossRef CAS PubMed.
- L. Qiao, Y. Lu, B. Liu and H. H. Girault, J. Am. Chem. Soc., 2011, 133, 19823–19831 CrossRef CAS PubMed.
- K. Yu, H. Zhang, J. He, R. N. Zare, Y. Wang, L. Li, N. Li, D. Zhang and J. Jiang, Anal. Chem., 2018, 90, 7154–7157 CrossRef CAS PubMed.
- J. Jiang, H. Zhang, M. Li, M. T. Dulay, A. J. Ingram, N. Li, H. You and R. N. Zare, Anal. Chem., 2015, 87, 8057–8062 CrossRef CAS PubMed.
- R. Qiu, X. Zhang, H. Luo and Y. Shao, Chem. Sci., 2016, 7, 6684–6688 RSC.
- W. Guo, H. Ding, C. Gu, Y. Liu, X. Jiang, B. Su and Y. Shao, J. Am. Chem. Soc., 2018, 140, 15904–15915 CrossRef CAS PubMed.
- J. L. Owens, H. A. Marsh and G. Dryhurst, J. Electroanal. Chem., 1978, 91, 231–247 CAS.
- Z. Jusys, H. Massong and H. Baltruschat, J. Electrochem. Soc., 1999, 146, 1093–1098 CrossRef CAS.
- O. Wolter and J. Heitbaum, Ext. Abstr., Meet. – Int. Soc. Electrochem., 1984, 6, 454–456 Search PubMed.
- O. Wolter and J. Heitbaum, Phys. Chem. Chem. Phys., 1984, 88, 6–10 CAS.
- A. A. Abd-El-Latif, C. J. Bondue, S. Ernst, M. Hegemann, J. K. Kaul, M. Khodayari, E. Mostafa, A. Stefanova and H. Baltruschat, TrAC, Trends Anal. Chem., 2015, 70, 4–13 CrossRef CAS.
- D. B. Trimarco, S. B. Scott, A. H. Thilsted, J. Y. Pan, T. Pedersen, O. Hansen, I. Chorkendorff and P. C. K. Vesborg, Electrochim. Acta, 2018, 268, 520–530 CrossRef CAS.
- M. Heinen, Y. X. Chen, Z. Jusys and R. J. Behm, Electrochim. Acta, 2007, 52, 5634–5643 CrossRef CAS.
- S. Brimaud, Z. Jusys and R. Jürgen Behm, Beilstein J. Nanotechnol., 2014, 5, 735–746 CrossRef CAS PubMed.
- J. Schnaidt, M. Heinen, Z. Jusys and R. J. Behm, J. Phys. Chem. C, 2012, 116, 2872–2883 CrossRef CAS.
- J. Schnaidt, M. Heinen, Z. Jusys and R. J. Behm, Catal. Today, 2013, 202, 154–162 CrossRef CAS.
- R. Reichert, J. Schnaidt, Z. Jusys and R. J. Behm, ChemPhysChem, 2013, 14, 3678–3681 CrossRef CAS PubMed.
- M. Gorlin, J. F. De Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- D. B. Trimarco, T. Pedersen, O. Hansen, I. Chorkendorff and P. C. K. Vesborg, Rev. Sci. Instrum., 2015, 86, 075006 CrossRef PubMed.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- A. Winiwarter, L. Silvioli, S. B. Scott, K. Enemark-Rasmussen, M. Sariç, D. B. Trimarco, P. C. K. Vesborg, P. G. Moses, I. E. L. Stephens, B. Seger, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2019, 12, 1055–1067 RSC.
- B. Liu, X. Y. Yu, Z. Zhu, X. Hua, L. Yang and Z. Wang, Lab Chip, 2014, 14, 855–859 RSC.
- Z. Wang, Y. Zhang, B. Liu, K. Wu, S. Thevuthasan, D. R. Baer, Z. Zhu, X. Y. Yu and F. Wang, Anal. Chem., 2017, 89, 960–965 CrossRef CAS PubMed.
- J. Yu, Y. Zhou, X. Hua, S. Liu, Z. Zhu and X. Y. Yu, Chem. Commun., 2016, 52, 10952–10955 RSC.
- T. C. Rohner, J. S. Rossier and H. H. Girault, Anal. Chem., 2001, 73, 5353–5357 CrossRef CAS PubMed.
- T. C. Rohner, J. S. Rossier and H. H. Girault, Electrochem. Commun., 2002, 4, 695–700 CrossRef CAS.
- G. J. Van Berkel, J. M. E. Quirke, R. A. Tigani, A. S. Dilley and T. R. Covey, Anal. Chem., 1998, 70, 1544–1554 CrossRef CAS PubMed.
- Á. I. López-Lorente, J. Izquierdo, C. Kranz and B. Mizaikoff, Vib. Spectrosc., 2017, 91, 147–156 CrossRef.
- W. Cheuquepán, J. M. Pérez, J. M. Orts and A. Rodes, J. Phys. Chem. C, 2014, 118, 19070–19084 CrossRef.
- M. Dunwell, X. Yang, Y. Yan and B. Xu, J. Phys. Chem. C, 2018, 122, 24658–24664 CrossRef CAS.
- A. Zimmermann, U. Künzelmann and L. Dunsch, Synth. Met., 1998, 93, 17–25 CrossRef CAS.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |