
Development of a unique reversible fluorescent probe for tracking endogenous sulfur dioxide and formaldehyde fluctuation in vivo†
Yanyan
Ma
 ,
Wenjie
Gao
,
Linlin
Zhu
,
Yuping
Zhao
and
Weiying
Lin
*
,
Wenjie
Gao
,
Linlin
Zhu
,
Yuping
Zhao
and
Weiying
Lin
*
Institute of Fluorescent Probes for Biological Imaging, School of Chemistry and Chemical Engineering, School of Materials Science and Engineering, University of Jinan, Jinan, Shandong 250022, P. R. China. E-mail: weiyinglin2013@163.com
First published on 20th August 2019
Abstract
A reversible fluorescent probe (NP) for sensing SO2 and FA was rationally constructed. With the outstanding attributes of NP, the fluctuation of endogenous SO2 and FA was successfully traced, not only at the cellular level, but also in living mice for the first time. Significantly, it was first found that the interaction of SO2 and FA can attenuate the cytotoxicity.
A living organism is a complex collection of diverse elements, ions and molecules which drive a web of interacting chemical reactions. Understanding the dynamic chemical interaction of these species offers both a challenge and an opportunity for further exploring the complex physiological processes. Sulfur dioxide (SO2), as an environmental pollutant and antioxidant, is often inhaled by human beings through the environment, food or pharmaceuticals.1,2 In living systems, it dissociates to its derivatives bisulfite and sulfite and has an important role in as a vasorelaxant in the cardiovascular system.3,4 It can also be metabolically generated from L-cysteine via the 3-mercaptopyruvate pathway or by the oxidation of thiosulfate in the presence of glutathione (GSH) and thiosulfate reductase.5,6 However, excessive SO2 production is associated with many neurological diseases7 and could cause vasodilation by upregulating the NO/cGMP signaling pathway.8,9 According to recent reports, formaldehyde (FA), which can be endogenously produced by methylation of DNA or histone demethylation, can trigger S-nitrosoglutathione (GSNO) reduction and deregulate the NO signaling pathway by mediating the S-protein nitrosylation.10 Furthermore, FA assimilation and enhancement in the protein S-nitrosation process could result in very low local GSH concentrations which is closely interrelated with the production of SO2 in living systems.11 Notably, SO2 and FA have a delicate relationship in the complex signal transduction pathways. However, up to now, the interrelated biological roles of SO2 and FA are still unclear. To explore the relationship between SO2 and FA in depth, visualization of these transient bio-species in a complex living organism is critical.
Fluorescence imaging has been widely used as a powerful approach to monitor physiologically active species in intact living samples because of its several advantages, such as high spatiotemporal resolution, high chemoselectivity and in situ real-time detection.12–16 Fluorescent probes can be divided into two categories: fluorescence intensity-based probes and ratiometric fluorescent probes. Compared to intensity-based probes, ratiometric fluorescent probes have attracted more attention due to their multichannel fluorescence signal output and the independence of the environmental effects, probe concentration and equipment efficiency.17–21 Recently, several excellent ratiometric fluorescent probes have been developed for SO2 detection.22–30 However, these irreversible probes are not capable of tracking the dynamic fluctuation of SO2 in real time. Although a few reversible fluorescent probes for monitoring of chalcogenides have been developed,31–33 a reversible fluorescence probe for tracking the dynamic fluctuation of endogenous SO2 and FA in vitro and in vivo has not been reported so far. To achieve this goal, the probes must have the merits of reversibility, fast response, deep penetration and high sensitivity, as well as a large signal variation. Therefore, the construction of the probes based on the previously mentioned attributes for the dynamic and reversible imaging of endogenous SO2 and FA in living cells and in vivo are highly desirable.
To address the previous issue, a reversible fluorescent probe NP was developed for monitoring the dynamic fluctuation between SO2 and FA. The overall design principle was inspired by two critical chemical reactions: (1) the Michael addition reaction, which is reversible; (2) FA is the main scavenger of bisulfite (Scheme S1, ESI†). In the preliminary work, a probe CP was constructed, which can promptly respond to SO2. However, an initial attempt to use CP for reversible visualizing of SO2 was unsuccessful.34 It was speculated that the electron withdrawing group (EWG) of the nearby carbonyl group may decrease the intrinsic reversibility of CP. Therefore, it was decided to achieve the reversibility by adjusting the EWG in the probe structure. In this work, the benzopyrylium unit was chose as both the energy acceptor and SO2 reaction site which has no EWG. Naphthalimide was selected as the energy donor due to its excellent photostability, high molar absorbance and high fluorescence quantum yield.35 Importantly, the fluorescence emission spectrum of the naphthalimide substantially overlaps with the absorption band of the flavylium derivative (Fig. S1, ESI†). Then piperazine was used to connect the donor and the acceptor together as the linker to develop a novel fluorescence resonance energy transfer (FRET)-based ratiometric fluorescence probe for sensing SO2 reversibility (Scheme 1). It was speculated that the FRET process may occur from the donor to the acceptor in the NP dyad, and the NP finally exhibited the red emission of the flavylium derivative. Upon addition of SO2, the conjugated system of benzopyrylium would be attacked, causing the prohibition of the FRET process and releasing the naphthalimide fluorescence. With the further addition of FA, the bisulfite group would be captured by FA to drive the dissociation of NP-HSO3 (the adduct NP and SO2) to form the probe NP. Simultaneously, the FRET process would be recovered in the NP dyad. Thus, the probe is reversible towards SO2 and FA. The synthesis steps and characterization data for the probe NP are presented in the ESI.†
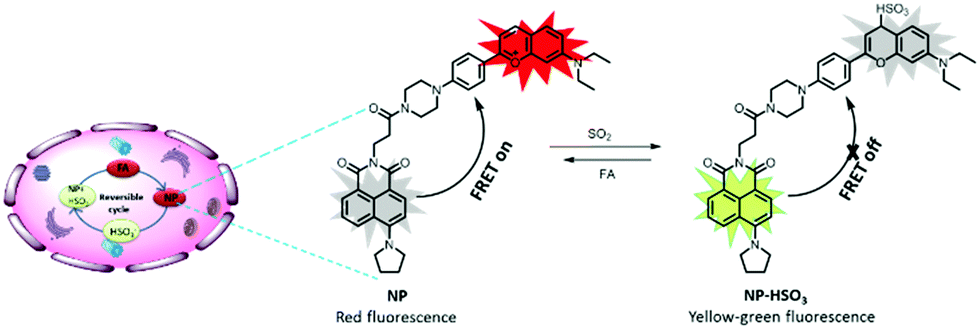 | ||
| Scheme 1 Rational design strategy and sensing mechanism of the probe NP. | ||
With the robust probe NP in hand, it was initially evaluated using titrimetric spectra analysis in PBS buffer. As shown in Fig. 1A, upon excitation at 446 nm, the free NP exhibited the emission band of the acceptor at 645 nm but no featured emission peak of naphthalimide donor, indicating that the FRET process from donor to acceptor existed. Whereas, with addition of an increasing concentration of NaHSO3 (a standard source for SO2), the fluorescence intensity at 645 nm of the acceptor gradually disappeared and a significant fluorescence emission of the donor at 540 nm increased. These results showed that the conjugation system of the acceptor was interrupted and that the FRET process had disappeared. Importantly, the ratio of fluorescence intensity (I540/I645) displayed a drastic variation from 0.015 to 4.8, a 320-fold increase which was highly desired. The sensing mechanism was confirmed by HRMS (Fig. S2, ESI†). In addition, the intensity ratio of I540/I645 exhibited an excellent linear relationship towards NaHSO3 concentrations ranging from 2 μM to 20 μM (Fig. 1B) with a detection limit of 0.86 μM, suggesting that the NP is potentially useful for detection of SO2.
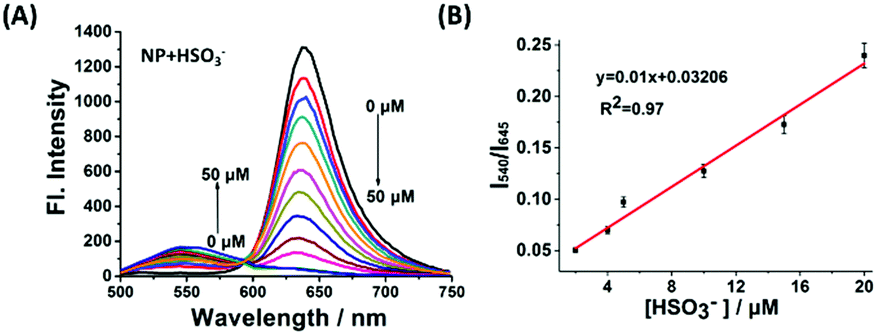 | ||
| Fig. 1 (A) The fluorescence responses of 10 μM NP upon addition of NaHSO3 (0–50 μM). λex = 446 nm. (B) The relationship between fluorescence ratio (I540/I645) and [HSO3−]. | ||
After verifying the suitability of NP for monitoring SO2, the reversibility of NP-HSO3 (the product of NP and SO2) was further investigated using FA. As shown in Fig. 2A, with an addition of various concentrations of FA to the solution (10 μM NP and 50 μM NaHSO3 in PBS buffer), an obvious fluorescence enhancement at 645 nm was obtained and the fluorescence emission at 540 nm was reduced simultaneously. The fluorescence intensity reached saturation after adding 200 μM FA, which is almost consistent with the fluorescence spectrum of NP. The variations of the absorption spectra of NP are consistent with the changes of fluorescence spectra (Fig. S3, ESI†). The ratio of I645/I540 recovered immediately in less than 150 s (Fig. S4, ESI†) and the reversible cycle could be repeated five times (Fig. 2B). The sensing mechanism was confirmed by HRMS (Fig. S5, ESI†). All the results demonstrated that NP-HSO3 can be effectively restored to NP by the addition of FA.
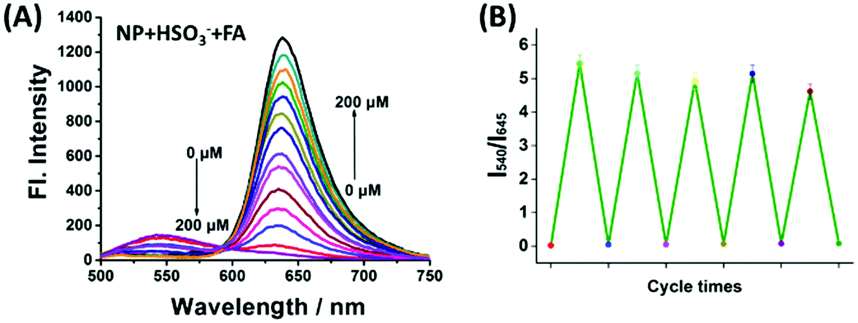 | ||
| Fig. 2 (A) The fluorescence spectra of NP-HSO3 (the product of 10 μM NP with 50 μM NaHSO3) in the presence of various concentrations of FA (0–200 μM). (B) Reversible cycle of NP upon alternate additions of NaHSO3 and FA. Error bars represent mean values ± SD (n = 3). | ||
The selectivity of NP towards SO2 was then explored. As shown in Fig. S6 (ESI†), NP exhibited obvious fluorescence ratio enhancement for SO2, whereas other bioactive small molecules did not cause obvious fluorescence alterations. In addition, the reversibility of NP-SO3H (10 μM NP treated with 50 μM NaHSO3) with other potential competing analytes was extensively investigated. As shown in Fig. S7 (ESI†), FA is the most effective species to restore the fluorescence of NP, up to an almost 95% recovery efficiency. By contrast, other analytes tested exhibited a minimum recovery efficiency. These data are indicative of the superior selectivity of the adduct NP-SO3H for FA over other species. Moreover, the NP displayed pH insensitivity (Fig. S8, ESI†). As shown in Fig. S9 (ESI†), the survival rate of HeLa cells after incubation with NP (0–50 μM) for 24 h was still over 85%, indicating that NP has a low cytotoxicity to living cells. The highly desirable attributes of the robust probe NP, including instantaneous response, reversibility and selectivity, make it possible to reversibly monitor the dynamic fluctuation of endogenous SO2 and FA in complex biological systems.
Initially, the imaging of exogenous SO2 and FA in living cells was investigated. As shown in Fig. S10 (ESI†), HeLa cells treated with NP (10 μM) mainly displayed a remarkable red fluorescence and a weak green fluorescence. Whereas upon addition of NaHSO3 (100 μM) to NP-loaded HeLa cells, an obvious fluorescence ratio (red/green) decrease was observed, indicating that SO2 interrupted the benzopyrylium π-conjugated system to form the NP-SO3H adduct. In addition, the fluorescence ratio was restored markedly after incubation with 200 μM FA, suggesting that the free bisulfite group is effectively captured by FA to drive the dissociation of the adduct NP-SO3H to the formation of the probe NP. Similar fluorescence changes could also be observed after alternate addition of NaHSO3 and FA and the reversible cycles could be repeated at least twice under the same conditions. These results indicate that the probe NP can track the dynamic interaction of exogenous SO2 and FA in living cells.
Recently, numerous reports have demonstrated that accumulation of FA is closely associated with several diseases.36 In the previous experiments, it was confirmed that exogenous SO2 and FA can interact with each other in the living cells, and then the biological influence of their interaction in the living cells was studied with fluorescence imaging, flow cytometry and Western blot analysis. As shown in Fig. S11 (ESI†), when the cells were treated with 1 mM FA for 24 h, a significant fluorescence enhancement in the blue channel was observed, suggesting that there was death of a great number of cells because of the adverse influence of FA. In sharp contrast, for the cells co-treated with 1 mM FA and various concentrations of NaHSO3 (0.5 and 1 mM) for 24 h, the fluorescence intensity in the blue channel was diminished with the increasing concentration of SO2, suggesting that SO2 can suppress FA-induced cytotoxicity. In addition, the MTT results were consistent with the results of the above fluorescence imaging (Fig. S12, ESI†).
To further confirm these findings, apoptosis experiments were carried out with flow cytometry and western blotting (Fig. 3). As shown in Fig. 3A, without the addition of SO2, FA would lead to more remarkable late apoptosis. For the cells incubated with 0.5 mM or 1 mM SO2 in the presence of 1 mM FA, the apoptosis was significantly reduced. Moreover, as seen from Fig. 3B, the changes in biochemical landmarks of apoptosis are consistent with the previously mentioned results. All the results confirmed that the interaction of SO2 and FA in the cells is able to block FA-induced cytotoxicity and there may be a certain of balance between SO2 and FA to maintain homeostasis. Therefore, providing a powerful chemical tool to explore the relationship between SO2 and FA in living systems in-depth is of great significance.
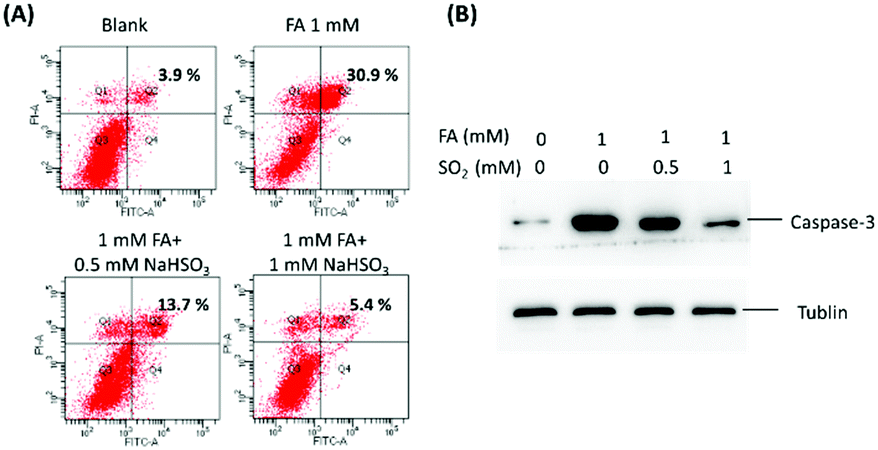 | ||
| Fig. 3 Biological influence of the interaction between SO2 and FA in U251 cells. (A) The apoptosis of U251 cells was assessed by flow cytometry. (B) The Western blot analysis of active caspase-3 in U251 cells. Tublin was used as a loading control. | ||
The robust applicability of NP for detecting endogenous SO2 and FA reversibly in living cells was then assessed. To achieve this goal, two stimulants were used: Cys, a key contributor to intracellular SO2 production,5 and tetrahydrofolate (Tet), which can be degraded to produce endogenous FA.37 As a control experiment, HeLa cells incubated with 200 μM Cys or 400 μM Tet exhibited no emission in red or green channels (Fig. S13, ESI†). However, HeLa cells incubated with 10 μM NP exhibited bright red fluorescence (Fig. 4). Upon addition of 200 μM Cys, a distinct decrease in fluorescence ratio (Fred/Fgreen) could be observed, indicating that SO2, the product of Cys metabolism, interrupted the conjugated system of the probe. Notably, upon further treatment with 400 μM Tet, the fluorescence ratio of HeLa cells was subsequently enhanced, suggesting that endogenous FA is produced and it can interact with endogenous SO2 to restore the fluorescence of the probe. Another reversible cycle can also be observed by successive addition of Cys and Tet. These data reveal, for the first time, that dynamic variation of endogenous SO2 and FA can be achieved in living cells by using the single reversible fluorescence probe NP.
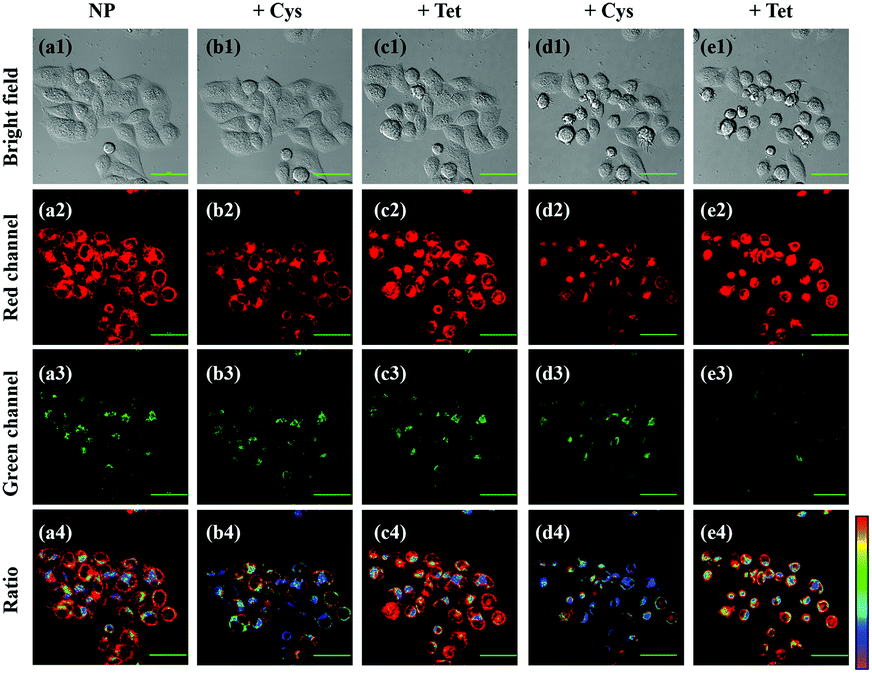 | ||
| Fig. 4 Reversible imaging of endogenous SO2 and FA by NP. (a1)–(e1) Bright-field images of HeLa cells continuously treated with 10 μM NP (a1)–(a4), 200 μM Cys (b1)–(b4), 400 μM Tet (c1)–(c4), 200 μM Cys (d1)–(d4) and 400 μM Tet (e1)–(e4) for 1 h. (a2)–(e2) Red channel: λex = 405 nm, λem = 663–738 nm. (a3)–(e3) Green channel: λex = 405 nm, λem = 500–550 nm. (a4)–(e4) Fluorescence ratio images of red channel and green channel. Scale bar: 20 μm. | ||
Based on the above desirable attributes, the applicability of NP in living mice was further evaluated using the favourable long-emission wavelength (645 nm) of NP. Reversible imaging of exogenous SO2 in living animals was carried out first. As shown in Fig. S14 (ESI†), the mice exhibited a weak fluorescence after treatment with only NP, whereas the mice displayed almost no fluorescence after intraperitoneal (i.p.) injection of NaHSO3. By contrast, after further injection of FA, the fluorescence intensity was progressively enhanced with increasing time and it reached saturation in 5 min. All of the data show that NP can detect exogenous SO2 and FA in living mice. It is noteworthy that the fluorescence intensity of living mice injected with FA was stronger than in the mice injected with only the probe, which implied that the probe may have potential for monitoring endogenous SO2 in living mice.
The ability of NP to reversibly image endogenous SO2 in the living mice was then examined. When injected (i.p.) with 50 μM NP, the mice showed weak fluorescence (Fig. S15, ESI†). Whereas, after injection of 2 mM FA into the previously NP-loaded mice, the red fluorescence was obviously enhanced with increasing time, validating for the first time that NP can be used to rapidly monitor endogenous SO2 in living mice.
Finally, the ability of NP for tracking the dynamic changes of endogenous SO2 and FA in the living mice was investigated (Fig. 5). The living mice exhibited weak fluorescence when injected with only NP. Upon injection of Tet, the fluorescence increased gradually, demonstrating that the endogenous FA was released by Tet and interacted with the endogenous SO2. Moreover, the fluorescence intensity was time-dependent and it attained saturation within 30 min. To further confirm that endogenous FA was produced by the Tet metabolism, NaHSO3, a scavenger of FA was injected into the above mice, and a significant fluorescence decrease was observed instantaneously. These data demonstrate for the first time that the unique probe can track the dynamic changes of endogenous SO2 and FA in living mice.
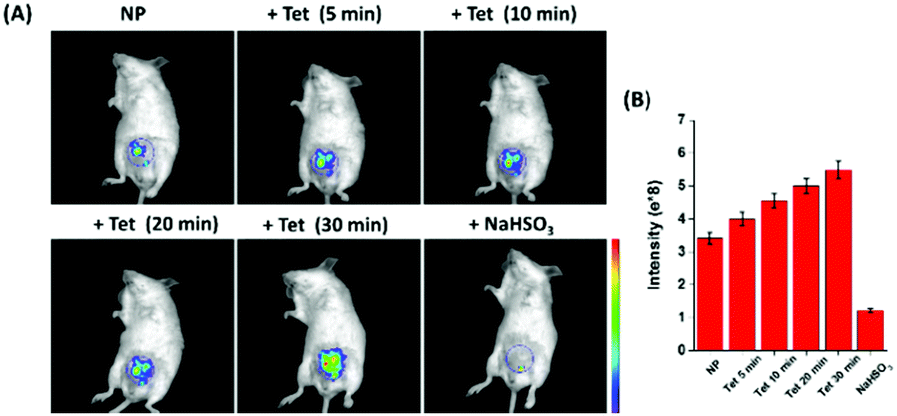 | ||
| Fig. 5 Fluorescence images of endogenous SO2 and FA in living mice. (A) Time-dependent fluorescence images of endogenous SO2 and FA in the living mice. (B) Quantification of fluorescence intensity in the living mice. | ||
In summary, a reversible fluorescent probe NP for tracking the dynamic changes of SO2in vitro and in vivo has been rationally designed and synthesized for the first time. The novel probe NP displayed highly desirable attributes, such as high selectivity, large fluorescence ratio change, and ultrafast response to SO2, and was effectively recovered by FA. For the first time, it was found that the interaction of SO2 and FA can protect U251 cells against apoptosis induced by FA. In addition, the new probe was successful used to reversibly monitor the dynamic changes of endogenous SO2 and FA in living cells and living mice for the first time, showing its powerful application. It is believed that the robust reversible probe NP can act as an effective chemical tool to further explore, in depth, the internal relationship between SO2 and FA and promote the initial research for SO2 and FA related diseases in biology and pathology.
This work was financially supported by the NSFC (21472067, 21672083, 21877048), the Taishan Scholar Foundation (TS201511041), and the start-up fund of the University of Jinan (309-10004).
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. C. Schroeter, J. Pharm. Sci., 2010, 50, 891–901 CrossRef.
- H. Vally and P. J. Thompson, Thorax, 2001, 56, 763–769 CrossRef CAS PubMed.
- X. Wang, H. Jin, C. Tang and J. Du, Eur. J. Pharmacol., 2011, 670, 1–6 CrossRef CAS PubMed.
- S. Du, H. Jin, D. Bu, X. Zhao, B. Geng, C. Tang and J. Du, Acta Pharmacol. Sin., 2010, 29, 923–930 CrossRef PubMed.
- M. H. Stipanuk and I. Ueki, J. Inherited Metab. Dis., 2011, 34, 17–32 CrossRef CAS PubMed.
- K.-H. Leung, G. B. Post and D. B. Menzel, Toxicol. Appl. Pharmacol., 1985, 77, 388–394 CrossRef CAS PubMed.
- M. Niu, Y. Han, Q. Li and J. Zhang, Neurosci. Lett., 2018, 665, 22–28 CrossRef CAS PubMed.
- J. Li, R. Li and Z. Meng, Eur. J. Pharmacol., 2010, 645, 143–150 CrossRef CAS PubMed.
- J. Li and Z. Meng, Nitric oxide, 2009, 20, 166–174 CrossRef CAS PubMed.
- C. M. Thompson, R. Ceder and R. C. Grafström, Toxicol. Lett., 2010, 193, 1–3 CrossRef CAS PubMed.
- C. A. Staab, J. Ålander, M. Brandt, J. Lengqvist, R. Morgenstern, R. C. Grafström and J.-O. Höög, Biochem. J., 2008, 413, 493–504 CrossRef CAS PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- J. Zhou and H. Ma, Chem. Sci., 2016, 7, 6309–6315 RSC.
- J. Zhu, C. Yu, Y. Chen, J. Shin, Q. Cao and J. Kim, Chem. Commun., 2017, 53, 4342–4345 RSC.
- L. X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2013, 114, 590–659 CrossRef PubMed.
- A. A. Kamnev, Spectrochim. Acta, Part A, 2018, 204, 576–580 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- L. Hee, K. Seung and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- W. Shi and H. Ma, Chem. Commun., 2012, 48, 8732–8744 RSC.
- L. He, B. Dong, Y. Liu and W. Lin, Chem. Soc. Rev., 2016, 45, 6449–6461 RSC.
- H. Chen, T. Sun, X. Qiao, Q. Tang, S. Zhao and Z. Zhou, Spectrochim. Acta, Part A, 2018, 204, 196–202 CrossRef CAS PubMed.
- G. Yin, Y. Gan, T. Yu, T. Niu, P. Yin, H. Chen, Y. Zhang, H. Li and S. Yao, Talanta, 2019, 191, 428–434 CrossRef CAS PubMed.
- W. Liu, D. Zhang, B. Ni, J. Li, H. Weng and Y. Ye, Sens. Actuators, B, 2019, 284, 330–336 CrossRef CAS.
- Y. Liu, J. Nie, W. Wang and W. Lin, J. Mater. Chem. B, 2018, 6, 1973–1983 RSC.
- M. Huang, L. Chen, J. Ning, W. Wu, X. He, J. Miao and B. Zhao, Sens. Actuators, B, 2018, 261, 196–202 CrossRef CAS.
- D. Li, Z. Wang, H. Su, J. Miao and B. Zhao, Chem. Commun., 2017, 53, 577–580 RSC.
- D. Zhang, A. Liu, R. Ji, J. Dong and Y. Ge, Anal. Chim. Acta, 2019, 1055, 133–139 CrossRef CAS PubMed.
- X. Yang, Y. Zhou, X. Zhang, S. Yang, Y. Chen, J. Guo and R. Yang, Chem. Commun., 2016, 52, 10289–10292 RSC.
- Y. Yue, F. Huo, P. Ning, Y. Zhang, J. Chao, X. Meng and C. Yin, J. Am. Chem. Soc., 2017, 139, 3181–3185 CrossRef CAS PubMed.
- Y. Zhao, Y. Ma and W. Lin, Sens. Actuators, B, 2019, 288, 519–526 CrossRef CAS.
- W. Zhang, T. Liu, F. Huo, P. Ning, X. Meng and C. Yin, Anal. Chem., 2017, 89, 8079–8083 CrossRef CAS PubMed.
- Z. Liu, S. Guo, J. Piao, X. Zhou and X. Wu, RSC Adv., 2014, 4, 54554–54557 RSC.
- S. Manjare, Y. Kim and D. Churchill, Acc. Chem. Res., 2014, 47, 102985 CrossRef PubMed.
- Y. Ma, Y. Tang, Y. Zhao, S. Gao and W. Lin, Anal. Chem., 2017, 89, 9388–9393 CrossRef CAS PubMed.
- P. A. Panchenko, O. A. Fedorova and Y. V. Fedorov, Russ. Chem. Rev., 2014, 83, 155 CrossRef.
- Z. Tong, C. Han, W. Luo, X. Wang, H. Li, H. Luo, J. Zhou, J. Qi and R. He, Age, 2013, 35, 583–596 CrossRef PubMed.
- G. Burgos-Barragan, N. Wit, J. Meiser, F. A. Dingler, M. Pietzke, L. Mulderrig, L. B. Pontel, I. V. Rosado, T. F. Brewer, R. L. Cordell, P. S. Monks, C. J. Chang, A. Vazquez and K. Patel, Nature, 2017, 548, 549–554 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc04411f |
| This journal is © The Royal Society of Chemistry 2019 |