
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sheath-flow probe electrospray ionization (sfPESI) mass spectrometry for the rapid forensic analysis of human body fluids†
Stephanie
Rankin-Turner
 *ac,
Satoshi
Ninomiya
b,
James C.
Reynolds
a and
Kenzo
Hiraoka
*c
*ac,
Satoshi
Ninomiya
b,
James C.
Reynolds
a and
Kenzo
Hiraoka
*c
aDepartment of Chemistry, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: s.rankin@lboro.ac.uk
bInterdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, 4-3-11, Takeda, Kofu, Yamanashi, 400-8511 Japan
cClean Energy Research Center, University of Yamanashi, 4-3-11, Takeda, Kofu, Yamanashi, 400-8511 Japan. E-mail: hiraoka@yamanashi.ac.jp
First published on 9th July 2019
Abstract
Sheath-flow probe electrospray ionisation (sfPESI) has for the first time been applied to the analysis of both fresh and dried human blood, saliva and urine. sfPESI enables the in situ sampling of biological materials with no sample preparation, demonstrating a promising technique for the rapid analysis and identification of body fluids of forensic interest.
Introduction
Biological materials recovered from crime scenes can offer vital information to a criminal investigation, particularly relating to the circumstances of the crime and the individuals involved. Not only can the presence of body fluids offer an insight into the incident, but the extraction of DNA from such material may be essential in identifying a victim or suspect. As the use of DNA profiling to aid legal investigations has become increasingly commonplace, the need to rapidly analyse and identify body fluids is more important than ever.A number of body fluids are commonly encountered at crime scenes, particularly blood, saliva, semen, urine, vaginal fluid and sweat. The process of identifying a suspected biofluid stain may involve a series of tests, often beginning with presumptive testing. Presumptive tests are frequently conducted in situ at a crime scene to establish the identity of a suspected biological sample, as a screening step before more complex tests such as DNA profiling are performed. Presumptive tests often involve the addition of a chemical reagent to the sample, resulting in a chemical or enzymatic reaction to indicate a positive result, typically via a colour change.1 For example, the addition of the reagent luminol to a blood sample results in luminescence caused by the oxidation of luminol in the presence of an oxidant such as hydrogen peroxide catalysed by heme.2 Common tests used for the detection of saliva are based on a reaction with amylase, an enzyme prevalent in the sample, whereas urine presumptive tests typically focus on the presence of urea and creatinine.1 Typically, there are specific presumptive tests required for each different body fluid and there is no established universal method for the simultaneous detection of multiple body fluids.
Unfortunately these presumptive tests are often non-specific, cross-reacting with specimens other than body fluids and thus resulting in false positive test results.3 Similarly, some tests lack specificity and will react with multiple body fluids or other species which may be present.1 As a result, further testing may be required following transportation of the sample to a laboratory, inevitably increasing the time taken to obtain a confirmatory identification of the body fluid. In addition to this, the presumptive tests commonly used are destructive, resulting in sample consumption or destruction of the DNA,4 rendering the sample useless for DNA analysis. This is a significant issue in a criminal investigation where the sample amount may be limited and preservation of evidence is often essential.
In recent years, there has been a push towards the application of analytical technologies to the identification of human body fluids. Studies have utilised a range of techniques including Raman spectroscopy,5,6 Fourier transform infrared (FTIR) spectroscopy,7–9 and mass spectrometry10–12 to identify body fluids. Mass spectrometry offers the possibility of detecting and identifying a wide range of low-level analytes in body fluids, but such methods frequently require destructive extraction and analysis steps. A suitable method for body fluid analysis would need to be specific enough to differentiate different biological materials, sufficiently sensitive to be applied to small sample sizes, and, particularly in the case of a forensic investigation, as non-destructive as possible. Furthermore, the ability to conduct rapid analysis without the need for sample pre-treatment would enable faster body fluid identification and potentially speed up a criminal investigation. Finally, it is essential that the method is capable of identifying both fresh and dried biological samples on a range of surface types, as those encountered in a real-world forensic scenario may have been present at the scene for days, weeks or even longer.
Mass spectrometry is already a gold standard in forensic analysis, particularly in the identification of illicit substances. Although traditionally a benchtop instrument, recent advances in ambient ionisation mass spectrometry have provided a potentially powerful range of tools for the rapid, in situ analysis of samples. Unfortunately these developments have not yet been utilised in the field of forensic body fluid identification. Since the introduction of the first ambient ionisation methods, desorption electrospray ionisation (DESI)13 and direct analysis in real time (DART),14 the range of techniques available has increased considerably,15 enabling direct analysis of samples in their native state without the requirement for prior sample preparation. The application of ambient ionisation techniques to biological materials has primarily focussed on the detection of specific compounds in body fluids, such as illicit and therapeutic drugs16,17 or biomarkers related to specific diseases.18–20 Surprisingly, ambient ionisation mass spectrometry has not been extensively utilised for the purpose of the forensic identification of biological materials.21
Probe electrospray ionisation (PESI) is an ambient ionisation technique developed by Hiraoka et al. which achieves electrospray from the tip of a solid needle.22 In brief, a needle tip is touched to the liquid surface of a sample, enabling the transfer of a small amount of sample solution to the needle tip. After this, the needle is raised until level with the mass spectrometer inlet, at which point a high voltage is applied. Upon application of a high voltage to the needle, an electrospray is generated, enabling rapid analysis whilst reducing or eliminating many issues associated with standard ESI techniques, including ion suppression and clogging of the capillary. Furthermore, PESI utilises an extremely small sample amount, estimated to be just a few picolitres.23 PESI has been successfully applied to a range of analytes, including illicit drugs in body fluids,24 biological tissues25,26 and food products.27 However as PESI is only applicable to liquid samples, sheath-flow PESI (sfPESI) was later developed to enable the analysis of non-liquid samples.28,29 Sheath-flow PESI is a modification of the PESI technique in which the needle is contained within a solvent-filled gel-loading tip with a slight protrusion (∼0.1 mm) of the needle from the base. When the probe touches the sample surface, the convex liquid at the base of the probe wets the sample and enables analyte extraction from the surface. Due to the small diameter of the probe tip, a minimal area of the sample surface is affected by the solvent extraction (approximately 1 mm2). The mechanism of sfPESI has been described in greater detail elsewhere.28
Here we present a sheath-flow PESI mass spectrometry technique capable of tackling many of the current downfalls of existing forensic body fluid identification techniques. This paper demonstrates the applicability of the method to both fresh and dried blood, saliva and urine, enabling the rapid analysis of complex biological samples, even after storage for up to 30 days.
Method
Probe construction
The sheath-flow PESI probe (Fig. 1) consists of a 0.12 mm outer diameter stainless steel acupuncture needle (Seirin, Shizuoka, Japan) inserted into a 20 μl gel-loading tip (epT.I.P.S, Eppendorf, Hamburg, Germany) filled with solvent. The acupuncture needle tip protruded from the base of the gel loading tip by approximately 0.1 mm. The needle was fixed into position with a silicone septum positioned at the top of the gel-loading tip.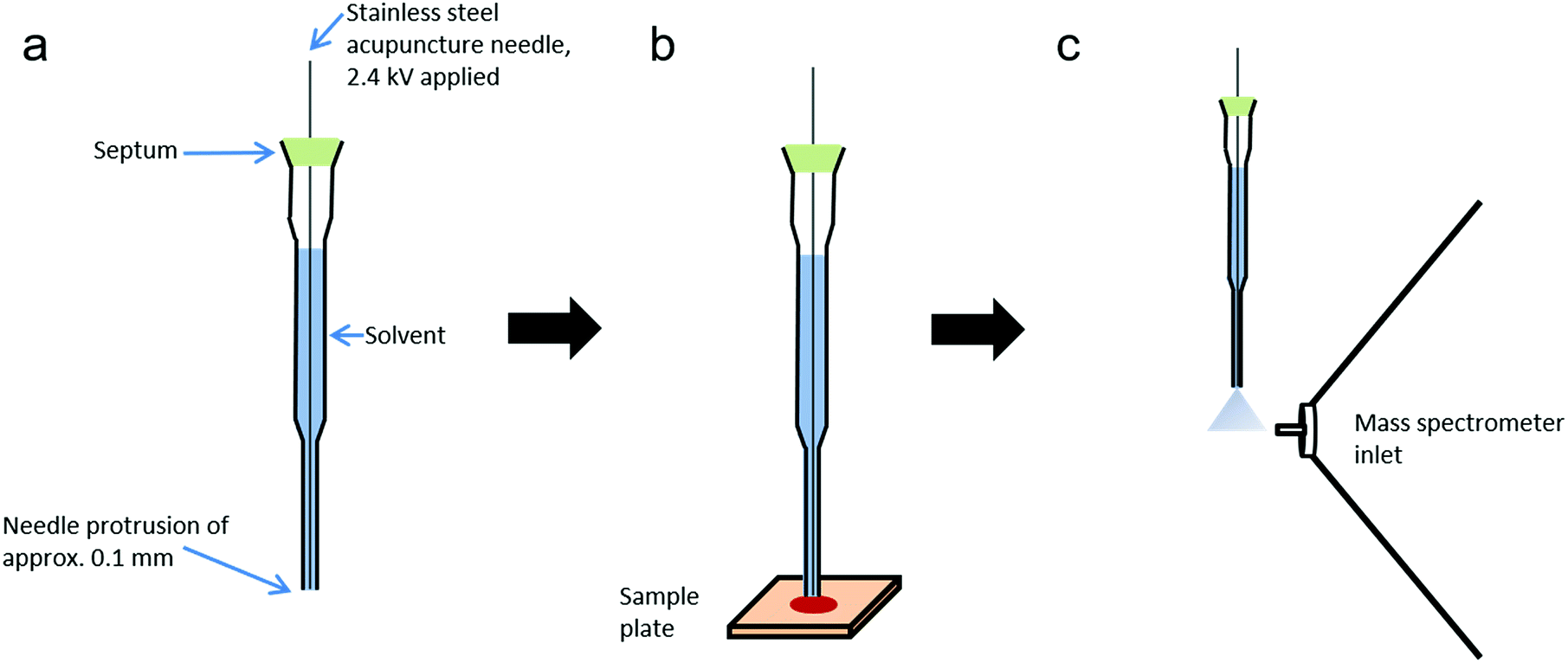 | ||
| Fig. 1 sfPESI schematic. (a) Construction of the sfPESI probe, (b) sfPESI probe briefly makes contact with sample to enable analyte extraction, and (c) probe is lifted to the mass spectrometer inlet and a high voltage applied to induce electrospray. | ||
Chemicals
The tip was filled with a sample extraction solvent of water/ethanol (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v). For Thermo Orbitrap MS experiments, purified water was obtained from a Simplicity UV (Millipore, Bedford, MA, USA) and 99.5% ethanol was purchased from Wako Pure Chemical Industries Ltd (Osaka, Japan). For Waters Synapt MS experiments, HPLC grade water was purchased from VWR (Lutterworth, UK) and 99.5% ethanol was purchased from Sigma-Aldrich (Gillingham, UK).
:50 v/v). For Thermo Orbitrap MS experiments, purified water was obtained from a Simplicity UV (Millipore, Bedford, MA, USA) and 99.5% ethanol was purchased from Wako Pure Chemical Industries Ltd (Osaka, Japan). For Waters Synapt MS experiments, HPLC grade water was purchased from VWR (Lutterworth, UK) and 99.5% ethanol was purchased from Sigma-Aldrich (Gillingham, UK).
Sampling
Samples were prepared by applying 5 μl of blood, saliva or urine to micro cover glass slides (Matsunami Glass, Osaka, Japan). Informed consent was obtained from the donor (Caucasian female) and procedures were approved by the ethical committee of the University of Yamanashi (No. 1872). Samples were either analysed immediately or stored under ambient conditions for analysis at a later date. Blood was also applied to aluminium foil, Whatman grade 1 chromatography paper, printer paper, cotton swabs and polyethylene plastic to assess the effects of surface type on analyte extraction.The tip of the probe was placed into contact with the sample surface for five seconds to enable sample extraction, during which time the probe was held at ground potential. The probe was then lifted to the highest position in front of the inlet of the mass spectrometer (2 mm above and 3 mm in front of the inlet) and 2.4 kV applied to the needle for five seconds by a high voltage power supply (Matsusada Precision, Shiga, Japan). The application of high voltage resulted in electrospray formation. All measurements were made using an Orbitrap mass spectrometer, with the exception of MS/MS experiments which were performed on a Waters Synapt mass spectrometer.
Orbitrap MS conditions
Measurements were made using an Orbitrap mass spectrometer (Exactive Plus, Thermo Fisher Scientific, San Jose, CA, USA) in positive ion mode with the following settings: m/z range 50–750, S-lens radiofrequency (RF) level 80, capillary temperature 100 °C, mass resolution 140000 and maximum ion injection time 100 ms. Data were acquired and analysed using Xcalibur software version 2.1 (Thermo Fisher Scientific, Bremen, Germany). For multivariate analysis, data were converted using MSConvert software (ProteoWizard 3.0)30 and input into MetaboAnalyst 4.0 for principal component analysis.31
Synapt MS conditions
Blood, saliva and urine samples were also analysed using a Waters Synapt high-definition mass spectrometer to perform MS/MS experiments for metabolite identification. The sfPESI probe was positioned in front of the mass spectrometer inlet and analysis performed in positive ion mode with the following mass spectrometer settings: sampling cone voltage 20 V, extraction cone voltage 3 V, source temperature 100 °C, transfer collision energy of 3 V and trap collision energy varied between 5 and 20 V. For Synapt experiments, high voltage was applied to the sfPESI needle by an adjustable 2.5 kV photomultiplier power supply, (Brandenburg, 476R model). Data were acquired and analysed using MassLynx version 4.1 software.Results and discussion
Sheath-flow probe electrospray ionisation was applied to the direct analysis of three human body fluids (blood, saliva and urine). In this study a 50:50 ethanol/water solvent mixture was used, however the probe solvent could be altered to facilitate the extraction of different types of compound depending on the analytes of interest. With a combined sample extraction and electrospray time of approximately 10 seconds, individual samples can be rapidly analysed, offering the possibility of high sample throughput.
Mass spectra recorded from the analysis of blood, saliva and urine demonstrated clearly distinct mass spectral profiles (Fig. 2). The collection of accurate mass data (<5 ppm mass error) enabled molecular formulae to be predicted and compound identities to be tentatively assigned based on knowledge of biofluid composition, the Human Metabolome Database,32 and previous literature.33–37 A range of compounds were detected in all body fluids, demonstrating the applicability of sfPESI to the detection of metabolites in biological materials. A select number of compounds of interest were further investigated by sfPESI-MS/MS to obtain fragmentation data to support the identifications made (see Table S1†). Spectra obtained from blood exhibited particularly strong signals at m/z 203 and 219, which have been assigned as sodiated and potassiated fructose/glucose respectively. At lower ion intensities, sodiated lipids were observed. A number of phospholipids (such as sodiated sphingomyelin at m/z 725) and cholesterol esters (such as sodiated and potassiated cholesteryl linoleate at m/z 671 and 687 respectively) could be observed in the higher m/z range.
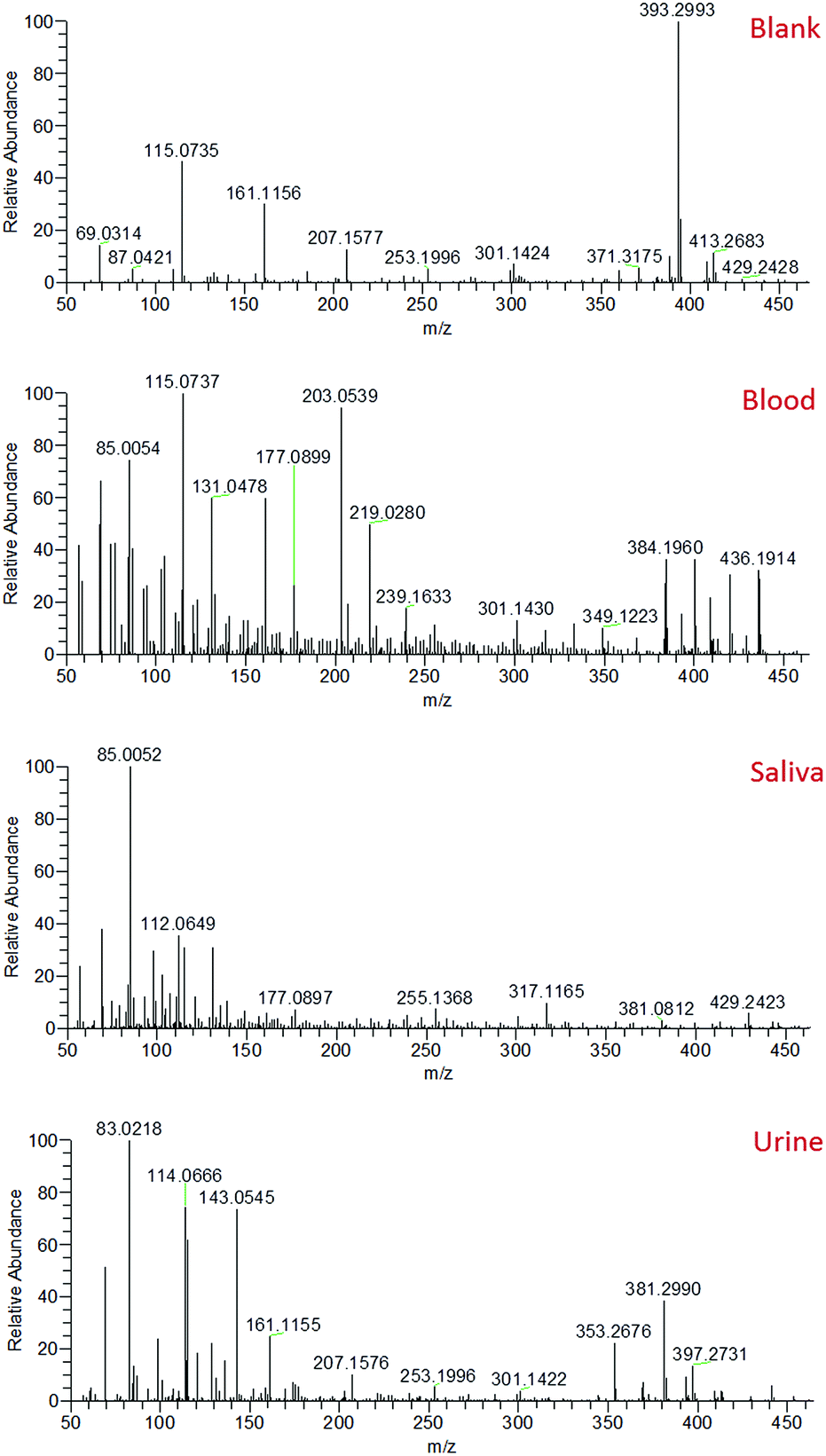 | ||
| Fig. 2 Typical mass spectra obtained from the solvent blank and fresh blood, saliva and urine. | ||
The most abundant ions observed in the saliva mass spectra actually pertain to compounds derived from the solvent. This may be a result of the viscosity of saliva affecting the amount of sample picked up by the sfPESI probe, or the lower concentrations at which analyte species are present in saliva compared to blood. Despite this effect, a number of protonated acids were identified, including 4-aminobutyric acid, 5-aminopentanoic acid, and methylimidazoleacetic acid at m/z 104, 118 and 141 respectively. Protonated amino acids could also be observed, including proline, threonine and phenylalanine at m/z 116, 120 and 166 respectively. The primary compounds detected in urine were, as would be expected, urea and creatinine (protonated, sodiated and both proton and sodium bound dimer species). These ions initially dominated the mass spectrum. The presence of many low-level metabolites (for example protonated creatine, proline betaine and 1-methylhistidine at m/z 132, 144 and 170 respectively) could also be detected, but the complete characterisation of all of these metabolites is beyond the scope of this paper. The presence of species such as urea and creatinine at high levels in urine would often require a pre-fractionation step such as solid phase extraction (SPE) or a chromatographic separation to enable urinary metabolites to be visualised. The sfPESI approach detailed here offers a novel solution to this issue enabling in situ analysis without the need for sample preparation. In standard ESI, the presence of surface-active compounds and charged components can result in the suppression of the signal from some analytes.38 Ion suppression can affect the accuracy and reliability of a method, and can even result in some analytes being completely lost. This is particularly problematic when dealing with complex matrices, such as biological samples. When Hiraoka et al. developed probe electrospray ionisation, a reduction in ion suppression was obtained due to the successive ionisation of analytes throughout the electrospray. More surface-active components were observed first, but when these are depleted in the sample, less surface-active components that typically may not have been observed due to ion suppression can be visualised.39
During the application of high voltage to the sfPESI probe, this same sequential ionisation can be observed in the urine sample. The technique demonstrates the possibility of a reduction in ion suppression, enabling the observation of other metabolites which would otherwise be masked by the creatinine and urea. Fig. 3 shows that the mass spectra obtained from fresh urine distinctly changes throughout the application of the high voltage. Initially (point T1), the mass spectrum is dominated by protonated and sodiated urea and creatinine (along with other adducts and dimers). However after a few seconds these compounds are depleted (point T2) and additional peaks corresponding to potential urinary metabolites can be observed (identified based on exact mass measurements and fragment ions where possible). This sequential ionisation is further demonstrated by Fig. S1,† which exhibits the depletion of creatinine throughout the electrospray followed by the appearance of an additional ion at m/z 218, assigned as protonated propionylcarnitine. The reproducibility of this is demonstrated in Fig. S2† (% RSD of repeated analyses 29.3% and 8.3% for creatinine and propionylcarnitine respectively), though reproducibility may be dependent on the sample and surface type. These data show that sfPESI may be capable of reducing in-source ionisation suppression of lower concentration analytes, which are only ionised upon the exhaustion of compounds such as urea and creatinine in the sample. Interestingly, sodiated ions are primarily only observed at point T1 of the sfPESI profile, which may indicate a depletion of salts throughout the sfPESI profile, resulting in protonation being the dominant ionisation mechanism for the species observed at point T2. This demonstrates the effectiveness of sfPESI to the in situ analysis of complex samples such as biological fluids, which often require some form of sample preparation and treatment prior to analysis.
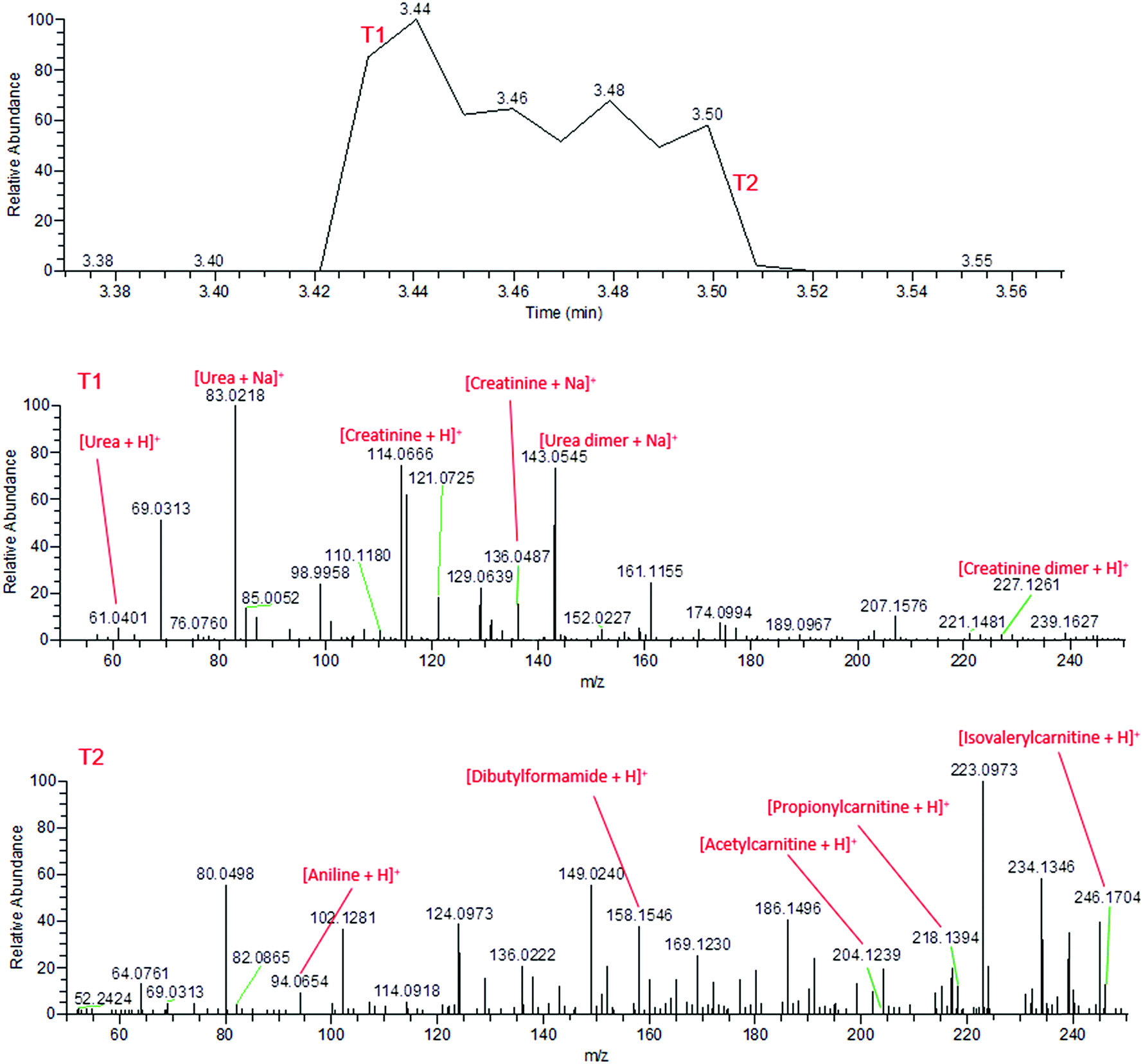 | ||
| Fig. 3 Mass spectra of fresh human urine, demonstrating the sequential ionisation from the beginning of the electrospray (T1, above) and the end of the electrospray (T2, below). | ||
By the time the application of high voltage comes to an end, the mass spectrum has returned to the signal observed from the solvent. This indicates the extracted sample is depleted within a matter of seconds, indicating negligible sample carryover.
Blood, saliva and urine samples were further analysed over a period of one month following storage under ambient conditions. Although sample analytes could still be detected at the one month time point on glass slides, chemical changes in the bodily fluid samples were observed. In the fresh human blood samples, the sodiated lipids in the m/z 650–750 region gradually decreased in intensity over the month of sample ageing, with some of these species disappearing completely from the mass spectrum by the one month time point (Fig. 4). For instance, sodiated sphingomyelin species at m/z 725 (identification supported by characteristic fragment ions observed at m/z 542 and 666) gradually decreased in intensity over time, though were still detectable after one month. This decrease may be due to the gradual decomposition of these compounds over time. Other prominent compounds were also found to decrease in abundance over time, such as sodiated and potassiated fructose/glucose. Alongside this there appeared to be an increase in the presence and abundance of lower mass compounds in all body fluids, indicating the formation of lower mass compounds as the biological fluid ages (Fig. S3†). This may be the result of the degradation of larger components into lower mass products, or the production of these species as a result of bacterial action on the surface of the sample.40,41 These data suggest that given further development sfPESI could potentially be used for the differentiation of biological stains of different ages, a problem that is currently a vital area of research in forensic science.42
 | ||
| Fig. 4 Decreasing signal intensity of phospholipids in blood over a period of one month. | ||
Finally, the suitability of sheath-flow PESI probe for biofluid analysis from different surface types was then investigated. In the context of a real-world criminal investigation, biological samples are frequently encountered on a variety of surfaces, such as clothing, objects and floors. It was therefore essential to investigate the plausibility of sampling directly from different surface types. To achieve this, fresh and dried blood was sampled directly from a number of surface materials – glass, plastic, cotton, foil, filter paper and printer paper. In order to assess the effects of the various matrices on sample detection, the observed intensity of an ion of interest was monitored (in this case, m/z 203.05, pertaining to sodiated fructose/glucose) when sampled from these different surface types.
Both fresh and dried (for approximately 2 hours) blood could be detected from all surface types, which included a range of both porous and non-porous surfaces. Although blood could be sampled from all materials tested, a notable difference in ion intensity was observed between porous (cotton and paper-based) and non-porous (foil, glass and plastic) surfaces. As would be expected, in all cases a decrease in analyte intensity was observed after sample drying. A greater decrease in analyte intensity after drying was observed with the porous surface materials (Fig. S4†), with analyte signal from the porous surfaces exhibiting an average decrease of 68% as opposed to an average decrease of 33% exhibited by non-porous materials. This effect is expected due to the absorption of the liquid sample into the porous material and greater dispersion of the sample, whereas the sample applied to non-porous materials remains on the surface. Furthermore, when the solvent meniscus formed at the tip of the sfPESI probe comes into contact with porous surfaces, some of the solvent is absorbed by the porous surface, thus resulting in a decreased analyte response. Fresh samples exhibited increased variability compared to dry samples, with samples deposited onto foil demonstrating the greatest variability between replicates. Despite this, the surface type experiment demonstrates the applicability of sfPESI to various surface materials, ensuring the technique could be applied to both fresh and dried body fluids encountered on a range of surface types often encountered in forensic investigations. Furthermore, separation between the three bodily fluids could be achieved in PCA regardless of surface type. Fig. 5 demonstrates the separation between blood, saliva and urine, with blood being sampled from a range of surface materials. The two samples outside of the 95% confidence region were fresh blood sampled from cotton. Fig. S5† displays the separation between body fluids when sampled from an identical surface. These data indicate the possibility of differentiating between different biological sample types regardless of the surface matrix.
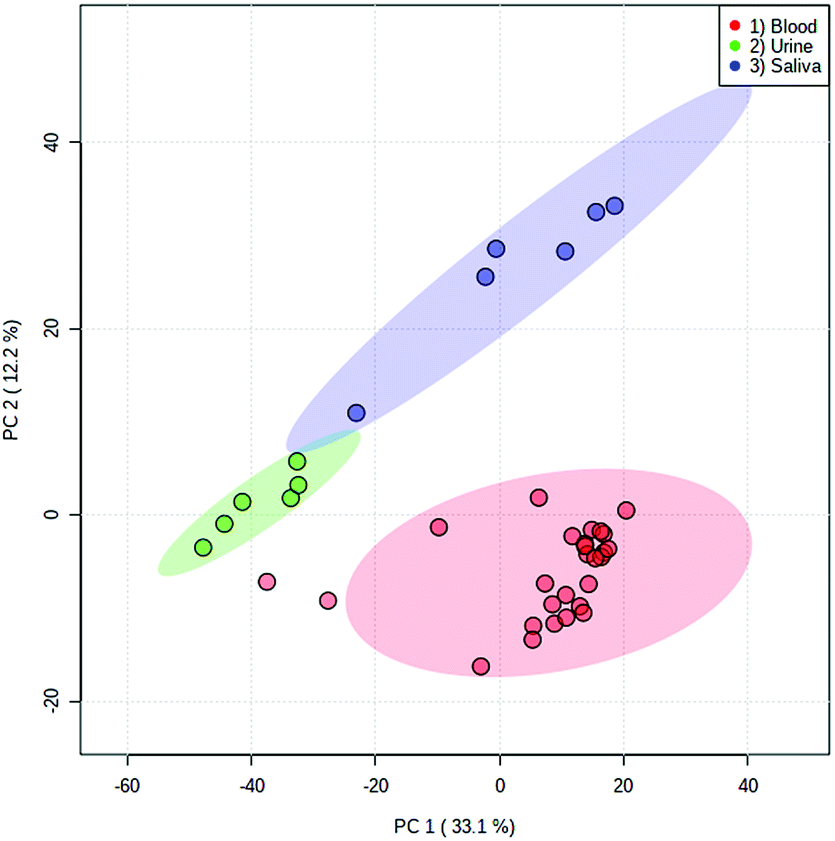 | ||
| Fig. 5 PCA scores plot of blood (red), urine (green) and saliva (blue) with log transformation and Pareto scaling. Urine and saliva were sampled from glass slides, whereas blood was sampled from a range of porous and non-porous surfaces. Ellipses depict 95% confidence regions. | ||
The processes involved in sfPESI share similarities with liquid extraction surface analysis (LESA), in which a small amount of solvent is applied to the surface of a sample for analyte extraction, after which the solvent is aspirated and electrospray initiated at a nano ESI chip positioned in front of the mass spectrometer.43 A recent study by Bailey et al.44 applied LESA-MS to the direct analysis of biological samples such as saliva and urine, demonstrating its ability to also simultaneously detect a wide range of metabolites. Although both techniques offer a rapid means of analyte extraction and analysis, sfPESI can allow for a more cost-effective approach, achieving analysis without the need to purchase the commercial LESA components. The amount of sample required in sfPESI has been estimated to be in the order of picograms, thus the technique can be used with minute sample amounts.28 Most importantly, the sequential ionisation observed with sfPESI enables the temporal separation of ions, offering the possibility of limiting ion suppression and visualising more analytes in such a way that has not been observed in other electrospray techniques such as LESA. The principles of this sequential ionisation have been described in greater detail in a recent paper by Usmanov et al.28
Conclusions
Sheath-flow probe electrospray ionisation mass spectrometry has been applied to the analysis of human body fluids for the purpose of forensic identification of suspected biological materials, specifically blood, saliva and urine. The technique was capable of analysing both fresh samples and dried samples stored for up to one month, demonstrating its applicability to body fluid analysis even after ageing and regardless of surface type. The sequential ionisation of different compounds observed demonstrates the ability of sheath-flow PESI to enable the observation of compounds that may have suffered from ion suppression effects using other ionisation techniques, thus improving confidence in identification of the biological sample. Furthermore, the use of this method revealed chemical changes occurring in the body fluids over time, indicating the possibility of identifying potential biomarkers of the age of a biological sample. In this study we have coupled sheath-flow PESI with a benchtop mass spectrometer, however the probe could be coupled with any portable mass spectrometer with an open inlet. Future research will explore the application of sfPESI with more portable analysers, opening up the possibility of truly portable in situ mass analysis. Furthermore, it will be necessary to confirm the ability to characterise biological fluids after the application of presumptive chemical reagents, as well as assessing the metabolic differences in biofluids from a greater number of participants. Validation of this method for body fluid identification would be achieved by applying multivariate analysis techniques to data obtained from a wide pool of donors. This rapid, preparation-free method of analysis shows great potential for use in the analysis of biological materials and could be readily applied to a range of fields of research given further development.Ethical statement
Informed consent was obtained from the donor who provided body fluid samples. Approval was obtained from the ethical committee of the University of Yamanashi (no. 1872), and the study was conducted in accordance with the ethical standards of the university and the Declaration of Helsinki.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the British Council and the Japan Society for the Promotion of Science (JSPS) via the JSPS Summer Program (SP18117).Notes and references
- K. Virkler and I. K. Lednev, Forensic Sci. Int., 2009, 188, 1–17 CrossRef CAS
.
- F. Barni, S. W. Lewis, A. Berti, G. M. Miskelly and G. Lago, Talanta, 2007, 72, 896–913 CrossRef CAS
- M. Vennemann, G. Scott, L. Curran, F. Bittner and S. S. Tobe, Forensic Sci., Med., Pathol., 2014, 10, 69–75 CrossRef CAS
- S. S. Tobe, N. Watson and N. N. Daéid, J. Forensic Sci., 2007, 52, 102–109 CrossRef CAS
- J. Fujihara, Y. Fujita, T. Yamamoto, N. Nishimoto, K. Kimura-Kataoka, S. Kurata, Y. Takinami, T. Yasuda and H. Takeshita, Int. J. Leg. Med., 2017, 131, 319–322 CrossRef CAS PubMed
- K. Virkler and I. K. Lednev, Forensic Sci. Int., 2008, 181, 1–5 CrossRef
- C.-M. Orphanou, Forensic Sci. Int., 2015, 252, 10–16 CrossRef
- K. M. Elkins, J. Forensic Sci., 2011, 56, 1580–1587 CrossRef CAS
- A. A. Quinn and K. M. Elkins, J. Forensic Sci., 2017, 62, 197–204 CrossRef CAS
- S. Kamanna, J. Henry, N. H. Voelcker, A. Linacre and K. P. Kirkbride, Int. J. Mass Spectrom., 2016, 397–398, 18–26 CrossRef CAS
- H. Yang, B. Zhou, H. Deng, M. Prinz and D. Siegel, Int. J. Leg. Med., 2013, 127, 1065–1077 CrossRef
- K. Van Steendam, M. De Ceuleneer, M. Dhaenens, D. Van Hoofstat and D. Deforce, Int. J. Leg. Med., 2013, 127, 287–298 CrossRef
- Z. Takáts, J. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef
- R. B. Cody, J. A. Laramée and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS
- R. Javanshad and A. R. Venter, Anal. Methods, 2017, 9, 4896–4907 RSC
- R. D. Espy, N. E. Manicke, Z. Ouyang and R. G. Cooks, Analyst, 2012, 137, 2344–2349 RSC
- T. J. Kauppila, N. Talaty, T. Kuuranne, T. Kotiaho, R. Kostiainen and R. G. Cooks, Analyst, 2007, 132, 868–875 RSC
- E. R. St John, M. Rossi, P. Pruski, A. Darzi and Z. Takats, TrAC, Trends Anal. Chem., 2016, 85, 2–9 CrossRef CAS
- K.-C. Schäfer, J. Dénes, K. Albrecht, T. Szaniszló, J. Balog, R. Skoumal, M. Katona, M. Tóth, L. Balogh and Z. Takáts, Angew. Chem., Int. Ed., 2009, 48, 8240–8242 CrossRef
- L. S. Eberlin, A. L. Dill, A. B. Costa, D. R. Ifa, L. Cheng, T. Masterson, M. Koch, T. L. Ratliff and R. G. Cooks, Anal. Chem., 2010, 82, 3430–3434 CrossRef CAS
- S. Rankin-Turner, M. A. Turner, P. F. Kelly, R. S. P. King and J. C. Reynolds, Chem. Sci., 2019, 10, 1064–1069 RSC
- K. Hiraoka, K. Nishidate, K. Mori, D. Asakawa and S. Suzuki, Rapid Commun. Mass Spectrom., 2007, 21, 3139–3144 CrossRef CAS
- K. Yoshimura, L. C. Chen, D. Asakawa, K. Hiraoka and S. Takeda, J. Mass Spectrom., 2009, 44, 978–985 CrossRef CAS
- S. Saha, M. K. Mandal and K. Hiraoka, Anal. Methods, 2013, 5, 4731 RSC
- K. Zaitsu, Y. Hayashi, T. Murata, K. Yokota, T. Ohara, M. Kusano, H. Tsuchihashi, T. Ishikawa, A. Ishii, K. Ogata and H. Tanihata, Anal. Chem., 2018, 90, 4695–4701 CrossRef CAS
- K. Yoshimura, L. C. Chen, M. K. Mandal, T. Nakazawa, Z. Yu, T. Uchiyama, H. Hori, K. Tanabe, T. Kubota, H. Fujii, R. Katoh, K. Hiraoka and S. Takeda, J. Am. Soc. Mass Spectrom., 2012, 23, 1741–1749 CrossRef CAS
- D. T. Usmanov, M. K. Mandal, K. Hiraoka, S. Ninomiya, H. Wada, M. Matsumura, S. Sanada-Morimura, H. Nonami and S. Yamabe, Food Chem., 2018, 260, 53–60 CrossRef CAS
- D. T. Usmanov, K. B. Ashurov, S. Ninomiya, K. Hiraoka, H. Wada, H. Nakano, M. Matsumura, S. Sanada-Morimura and H. Nonami, Rapid Commun. Mass Spectrom., 2018, 32, 407–413 CrossRef CAS
- K. Hiraoka, S. Rankin-Turner, S. Ninomiya, H. Wada, H. Nakano, M. Matsumura, S. Sanada-Morimura, F. Tanaka and H. Nonami, J. Agric. Food Chem., 2019, 67, 3275–3283 CrossRef CAS
- J. D. Holman, D. L. Tabb and P. Mallick, Curr. Protoc. Bioinf., 2014, 46, 1–9 Search PubMed
- J. Chong, O. Soufan, C. Li, I. Caraus, S. Li, G. Bourque, D. S. Wishart and J. Xia, Nucleic Acids Res., 2018, 46, 486–494 CrossRef
- D. S. Wishart, T. Jewison, A. C. Guo, M. Wilson, C. Knox, Y. Liu, Y. Djoumbou, R. Mandal, F. Aziat, E. Dong, S. Bouatra, I. Sinelnikov, D. Arndt, J. Xia, P. Liu, F. Yallou, T. Bjorndahl, R. Perez-Pineiro, R. Eisner, F. Allen, V. Neveu, R. Greiner and A. Scalbert, Nucleic Acids Res., 2013, 41, 801–807 CrossRef
- A. J. Lloyd, M. Beckmann, G. Favé, J. C. Mathers and J. Draper, Br. J. Nutr., 2011, 106, 812–824 CrossRef CAS
- C. R. Roe, D. S. Millington, D. A. Maltby, T. P. Bohan and C. L. Hoppel, J. Clin. Invest., 1984, 73, 1785–1788 CrossRef CAS
- Y. Zhu, P. S. H. Wong, M. Cregor, J. F. Gitzen, L. A. Coury and P. T. Kissinger, Rapid Commun. Mass Spectrom., 2000, 14, 1695–1700 CrossRef CAS
- F.-F. Hsu and J. Turk, J. Am. Soc. Mass Spectrom., 2000, 11, 437–449 CrossRef CAS
- P. M. Hutchins, E. E. Moore and R. C. Murphy, J. Lipid Res., 2011, 52, 2070–2083 CrossRef CAS
- C. G. Enke, Anal. Chem., 1997, 69, 4885–4893 CrossRef CAS
- M. K. Mandal, L. C. Chen and K. Hiraoka, J. Am. Soc. Mass Spectrom., 2011, 22, 1493–1500 CrossRef CAS
- E. J. Saude and B. D. Sykes, Metabolomics, 2007, 3, 19–27 CrossRef CAS
- S. Esfahani, N. M. Sagar, I. Kyrou, E. Mozdiak, N. O'Connell, C. Nwokolo, K. D. Bardhan, R. P. Arasaradnam and J. A. Covington, Biosensors, 2016, 6, 1–11 CrossRef
- R. H. Bremmer, K. G. De Bruin, M. J. C. Van Gemert, T. G. Van Leeuwen and M. C. G. Aalders, Forensic Sci. Int., 2012, 216, 1–11 CrossRef CAS
- V. Kertesz and G. J. Van Berkel, J. Mass Spectrom., 2010, 45, 252–260 CrossRef CAS
- M. J. Bailey, E. C. Randall, C. Costa, T. L. Salter, A. M. Race, M. de Puit, M. Koeberg, M. Baumert and J. Bunch, Anal. Methods, 2016, 8, 3373–3382 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay00698b |
| This journal is © The Royal Society of Chemistry 2019 |