
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature decarboxylative cyanation of carboxylic acids using photoredox catalysis and cyanobenziodoxolones: a divergent mechanism compared to alkynylation†
Franck
Le Vaillant
,
Matthew D.
Wodrich
 and
Jérôme
Waser
*
and
Jérôme
Waser
*
Laboratory of Catalysis and Organic Synthesis, Institut des Sciences et Ingénierie Chimiques, École polytechnique fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: jerome.waser@epfl.ch
First published on 22nd December 2016
Abstract
The one-step conversion of aliphatic carboxylic acids to the corresponding nitriles has been accomplished via the merger of visible light mediated photoredox and cyanobenziodoxolones (CBX) reagents. The reaction proceeded in high yields with natural and non-natural α-amino and α-oxy acids, affording a broad scope of nitriles with excellent tolerance of the substituents in the α position. The direct cyanation of dipeptides and drug precursors was also achieved. The mechanism of the decarboxylative cyanation was investigated both computationally and experimentally and compared with the previously developed alkynylation reaction. Alkynylation was found to favor direct radical addition, whereas further oxidation by CBX to a carbocation and cyanide addition appeared more favorable for cyanation. A concerted mechanism is proposed for the reaction of radicals with EBX reagents, in contrast to the usually assumed addition elimination process.
1. Introduction
Nitriles are extremely useful building blocks in organic synthesis and material science, especially in the synthesis of nitrogen containing heterocycles.1 Aliphatic nitriles in particular have found various applications in the fine chemicals industry, both as building blocks and final products in natural and synthetic bioactive compounds.1b,2 For example, anastrozole (1) is a blockbuster developed by AstraZeneca and is the drug of choice for treating breast cancer.3a Saxagliptin (2) is a classic drug for treating diabetes.3b Odanacatib (3), which is currently being developed by Merck, is expected to be a top selling drug for osteoporosis and bone metastasis in the upcoming years (Fig. 1).3c Nitriles often exhibit some bioactivity as bio-isosteres of carbonyl, halogens or others pharmacophores.2a Furthermore, the corresponding tetrazoles obtained after [3 + 2] cycloaddition with azides4 are also considered as bio-isosteres of the carboxylic acid group.5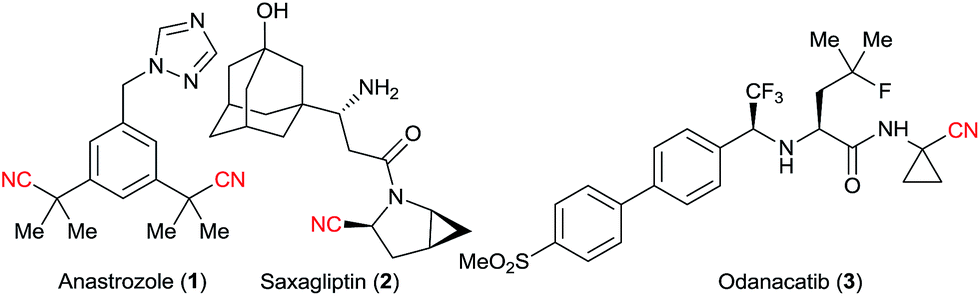 | ||
| Fig. 1 Importance of aliphatic nitriles in pharmaceuticals. | ||
Considering their importance, it is not surprising that many methods for the installation of nitriles have been developed (Scheme 1).1 Nucleophilic substitutions on primary alkyl halides are well established (Kolbe nitrile synthesis, Scheme 1A).6 However, side reactions like eliminations can proceed easily under these conditions. Furthermore, the alkyl halide precursors must first be synthetized. To overcome this limitation, C–H activation has recently received growing interest (Scheme 1B). However, it often encounters selectivity issues, needs high catalyst loading, and is limited to Csp2 carbons (using Rh, Co, Cu or Fe catalysts)7 or weak Csp3–H bonds, especially α to heteroatoms (using metal catalysts,8 or Hydrogen Atom Transfer (HAT)/oxidative methods).9
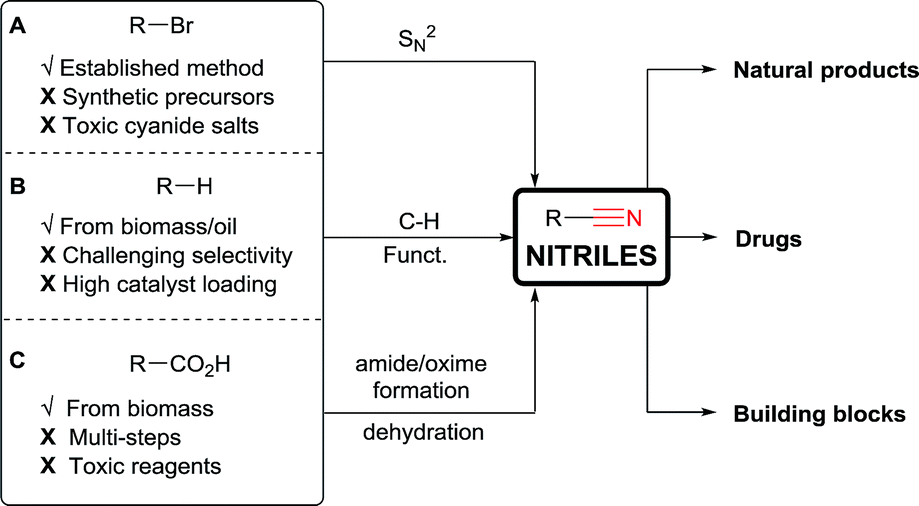 | ||
| Scheme 1 Applications and classical methods for nitriles synthesis. | ||
As broadly available substrates, carboxylic acids are attractive starting materials (Scheme 1C). Indeed, in nature, nitriles are synthesized through an enzymatic cascade starting from α-amino acids via a decarboxylative formation of aldoximes followed by dehydration.10 In synthetic chemistry, carboxylic acids have also been used to access nitriles. However, classical methods also involve multi-step procedures via the formation of amides or oximes followed by dehydration.11 Consequently, more efficient single step methods for the conversion of carboxylic acids to nitriles are needed.
In principle, a direct carboxylic acids–nitrile exchange would be a very efficient approach. However, this reaction occurs only at very high temperature (Scheme 2A).12 It was optimized by Klein in 1971 by heating carboxylic acids at 285 °C in the presence of α-methylglutaronitrile and phosphoric acid.12a This method has been applied in continuous flow by Kappe and Cantillo in 2013.12b An approach allowing milder reaction conditions is based on the radical decarboxylation of carboxylic acids followed by trapping of the in situ generated nucleophilic radical with a cyanation reagent. Barton and co-workers have developed a two steps visible light promoted decarboxylation via N-hydroxy-2-thiopyridone esters – the so called “Barton Esters” (Scheme 2B).13 Different reagents have been used to perform the cyanation of radicals, such as tosyl cyanide and organophosphoryl cyanides.13,14 Nevertheless, in this approach activation of the acids as Barton esters is required, leading to an additional synthetic step. A one-step decarboxylative cyanation of broadly available carboxylic acids would be therefore of high interest.
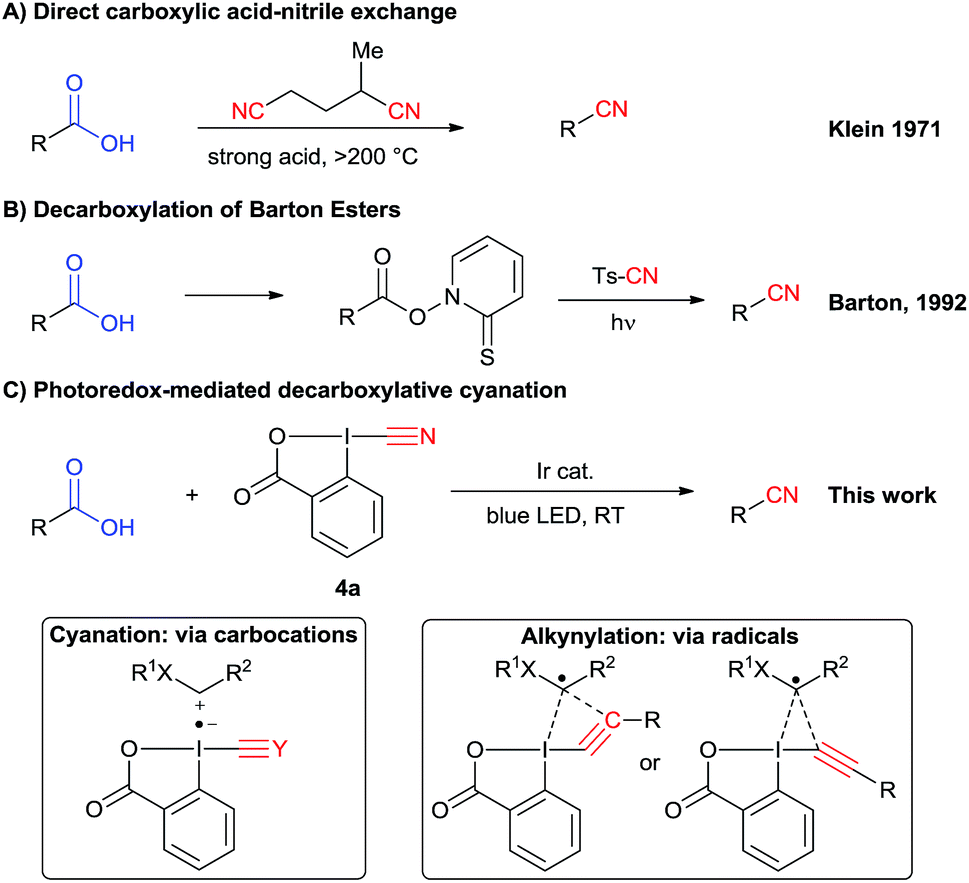 | ||
| Scheme 2 More efficient methods to convert carboxylic acids to nitriles. | ||
In this regard, visible light mediated catalysis has emerged as a powerful method for generating radicals with high chemoselectivity under mild conditions.15 In 2011, Rueping and co-workers reported a photoredox mediated oxidative Strecker reaction of tertiary amines using an iridium catalyst.16 In 2016, Opatz and co-workers were able to use an organic photocatalyst to promote this reaction.17 The same year, Xu and coworkers developed a cyanation of potassium alkyltrifluoroborates via photoredox catalysis using tosyl cyanide.18 The scope is limited to hydrocarbon-derived borates, and an excess of external oxidant and TFA is required. Recently, efficient photoredox-catalyzed decarboxylative transformations of carboxylic acids have been reported.19 In particular, the merger of photoredox catalysis and hypervalent iodine reagents for the decarboxylative alkynylation of aliphatic acids has been successfully and independently described by our group and the Xiao group.20 Key for success in this transformation was the use of ethynylbenziodoxolones (EBX reagents).
The corresponding cyanobenziodoxolone (CBX) reagent 4a was synthesized by Zhdankin and co-workers and used in the C–H cyanation of dialkylaryl amines.21 A radical pathway is probable for this transformation. Since then, cyanobenziodoxolones have successfully been used in the cyanation of nucleophiles,22 but have not yet been used in decarboxylative cyanation.23 Based on Zhdankin thermal cyanation with CBX and our previous decarboxylative alkynylation using EBX reagents and photoredox catalysis, we envisioned that CBX derivatives could be suitable reagents for the photoredox mediated cyanation of aliphatic acids using commercially available blue LEDs.
Herein, we report the successful implementation of this strategy using an iridium photoredox catalyst (Scheme 2C). The scope of the decarboxylative cyanation is broad, allowing the functionalization of various α-amino and α-oxy acids. Valuable intermediates in the synthesis of drugs have been synthetized in good yield. Dipeptides are also suitable for this transformation. Finally, we investigated the mechanism of both the previously developed alkynylation and the new cyanation. Based on experimental and computational data, we proposed different mechanisms for the two reactions, involving radical or carbocationic intermediates for alkynylation and cyanation, respectively. In the case of the alkynylation reaction, we further challenge the commonly accepted addition–elimination mechanism and propose that a concerted mechanism may be competitive.
2. Results and discussion
Optimization of the decarboxylative cyanation
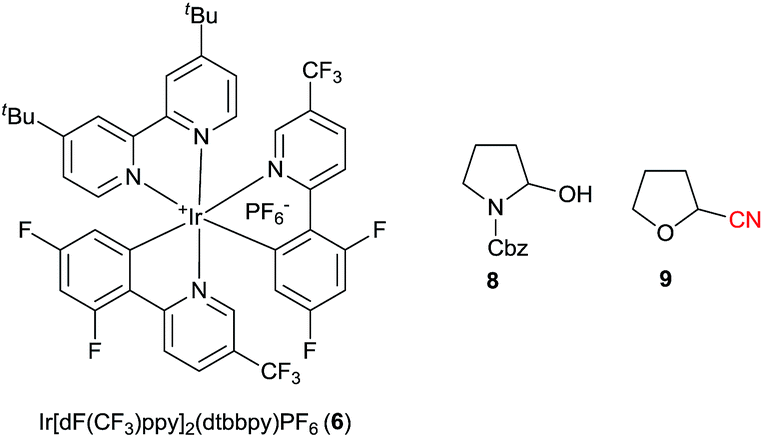
We began our investigations with the decarboxylative cyanation of protected proline 5a using the same conditions as we had reported for alkyne transfer (with 1 mol% Ir[dF(CF3)ppy]2(dtbbpy)PF6 (6), 3 equivalents CsOBz at room temperature in DCE, Table 1).20a,24 We were pleased to isolate 40% of the desired nitrile 7a after 4.5 h (entry 1). The moderate yield was mostly due to the formation of alcohol 8 as a side product. The origin of the oxygen atom could be either dioxygen or water, but as the reaction was done in degassed DCE, we speculated that the most probable source was water. Indeed, the formation of alcohol 8 could be suppressed by the addition of 4 Å molecular sieves. Together with a lower amount of cesium benzoate (1.5 equiv. instead of 3.0 equiv.), this led to an improvement of yield (78%) as well as reproducibility (entry 2). Decreasing the concentration to 0.05 M led to a decrease in yield to 46% (entry 3), while a concentration of 0.10 M afforded 72% of 8 (entry 4). Solubility issues started to be significant at 0.33 M, resulting in a lower yield (48%) (entry 5). A strong effect of the solvent was also observed: highly polar solvent such as DMF or DMSO led only to decomposition, while acetonitrile and toluene allowed the reaction to proceed, but only very slowly (entries 6 and 7). Performing the reaction in DCM did not affect the yield (entry 8). Finally, cyclic ethers such as THF and 1,4-dioxane were found to be the best solvents for this transformation (87 and 84% respectively, entries 9 and 10). Although small amounts of α-cyano-THF (9) could be isolated at the end of the reaction, the use of THF as solvent gave reproducibly better results. It is noteworthy that CsOBz was again the best base for this transformation, and especially CsOAc and Cs2CO3 did not lead to formation of the product (results not shown).|
|
||||
|---|---|---|---|---|
| Entry | Concentration (M) | Solvent | Conversiona (%) | Yieldb (%) |
| a Reaction conditions: 0.10 mmol 5a (1 equiv.), 0.15 mmol 4a (1.5 equiv.), 1 μmol 6 (0.01 equiv.) in DCE (0.5 mL) for 4.5 h at RT. The conversion of 5a by NMR is given. b Isolated yield after preparative TLC. c Same conditions used as for the decarboxylative alkynylation (3.0 equiv. CsOBz, no MS). d Solubility issue at 0.33 M. | ||||
| 1c | 0.20 | DCE | >95 | 40 |
| 2 | 0.20 | DCE | >95 | 78 |
| 3 | 0.05 | DCE | 90 | 46 |
| 4 | 0.10 | DCE | >95 | 72 |
| 5d | 0.33 | DCE | >95 | 48 |
| 6 | 0.20 | DMF/DMSO | >95 | Decomp. |
| 7 | 0.20 | MeCN/toluene | Low | Not isolated |
| 8 | 0.20 | DCM | >95 | 75 |
| 9 | 0.20 | THF | >95 | 87 |
| 10 | 0.10d | Dioxane | >95 | 84 |
|
||||

The structure of the reagent is important, particularly the core of the five-membered ring: 1-cyano-3,3-dimethyl-1,2-benziodoxole (CDBX, 4b) did not promote formation of 7a, instead generating only THF 2-carbonitrile (9) (Scheme 3). Electron withdrawing groups in para position to the iodine (CBX 4c and 4d) led to lower yields. The reaction was not complete with 4d, maybe due to the poorer solubility of this reagent. More electron-rich reagent 4e afforded a lower yield. Acyclic iodine reagent 4f led to no cyanation. Under these conditions, BrCN and ICN gave the product in less than 10%, and no reactivity was observed using tosyl cyanide. Only decomposition is observed when KCN is used. These results showed the superiority of benziodoxolone reagents as a cyanide source. Furthermore, CBX (4a) is a user friendly reagent, as it is a crystalline solid with a high melting point.
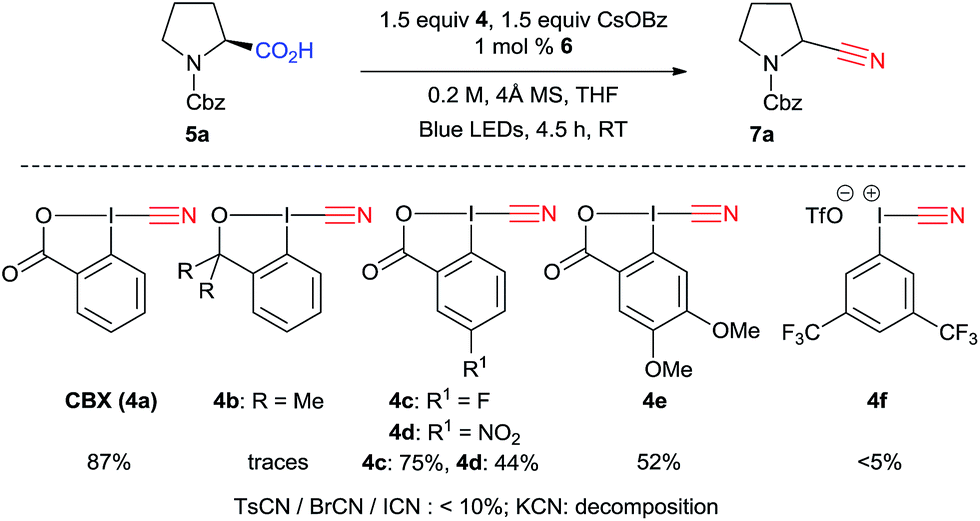 | ||
| Scheme 3 Screening of cyanation reagents. | ||
Investigation of the reaction scope
We then turned our attention to the scope of the reaction with amino acids (Scheme 4). When the reaction was scaled up from 0.10 mmol to 0.30 mmol, nitrile 7a was obtained in 89% yield. In this case, 9% of side product 9 was also observed.25 While in the previously developed alkynylation the reaction was sensitive to the substituent in α position of the amino acids, giving broadly varying yields, the decarboxylative cyanation is more general. In fact, both natural and unnatural α-amino acids can be functionalized in good yield in 5 to 18 hours under mild conditions. Different protecting groups, such as Cbz, Boc and Fmoc could be used, and cyanated proline derivatives 7a–c were obtained in excellent yield (86–92%). In the case of a less electron-withdrawing benzyl protecting group, cyanation still occurred, but only in 43% yield (product 7d). A free alcohol was tolerated to give 3-hydroxy proline derivatives 7e in 90% yield. Boc-protected piperidine 5f can be cyanated in 72% yield (product 7f). The reaction of Cbz-protected tetrahydroisoquinoline 3-carboxylic acid (5g) was site selective, yielding 65% of a single regioisomer 7g. From non-cyclic amino acids, primary, secondary and tertiary α-amino radicals can be generated and cyanated smoothly to furnish the corresponding nitriles 7h–j, although the yield is lower with tertiary radicals (51% for 7j). Valine, leucine and phenylalanine (5k–m) are suitable substrates (products 7k–m, 78–82%). For secondary radicals, the steric in α position did not strongly influence the reaction outcome. A benzyl ether was also tolerated in the transformation (product 7n, 80%). Protected glutamate, methionine and lysine 5o–q can be converted into the corresponding nitriles 7o–q in good to excellent yields (59–83%). The fact that the decarboxylative cyanation worked on methionine is especially noteworthy, as electrophilic cyanation reagents such as cyanogen bromide are known to react with this amino acid.26 Two dipeptides (Z-Gly-Pro-OH (5r) and Z-D-Phe-Pro-OH (5s)) could also be cyanated (products 7r and 7s). Cyanide 7s was obtained as a mixture of diastereoisomers. These preliminary results are promising for the cyanation of more complex amino acids. On the other hand, the reaction was not successful for tryptophan derivatives or when a sulfur atom was present in the β position (products 7t and 7u).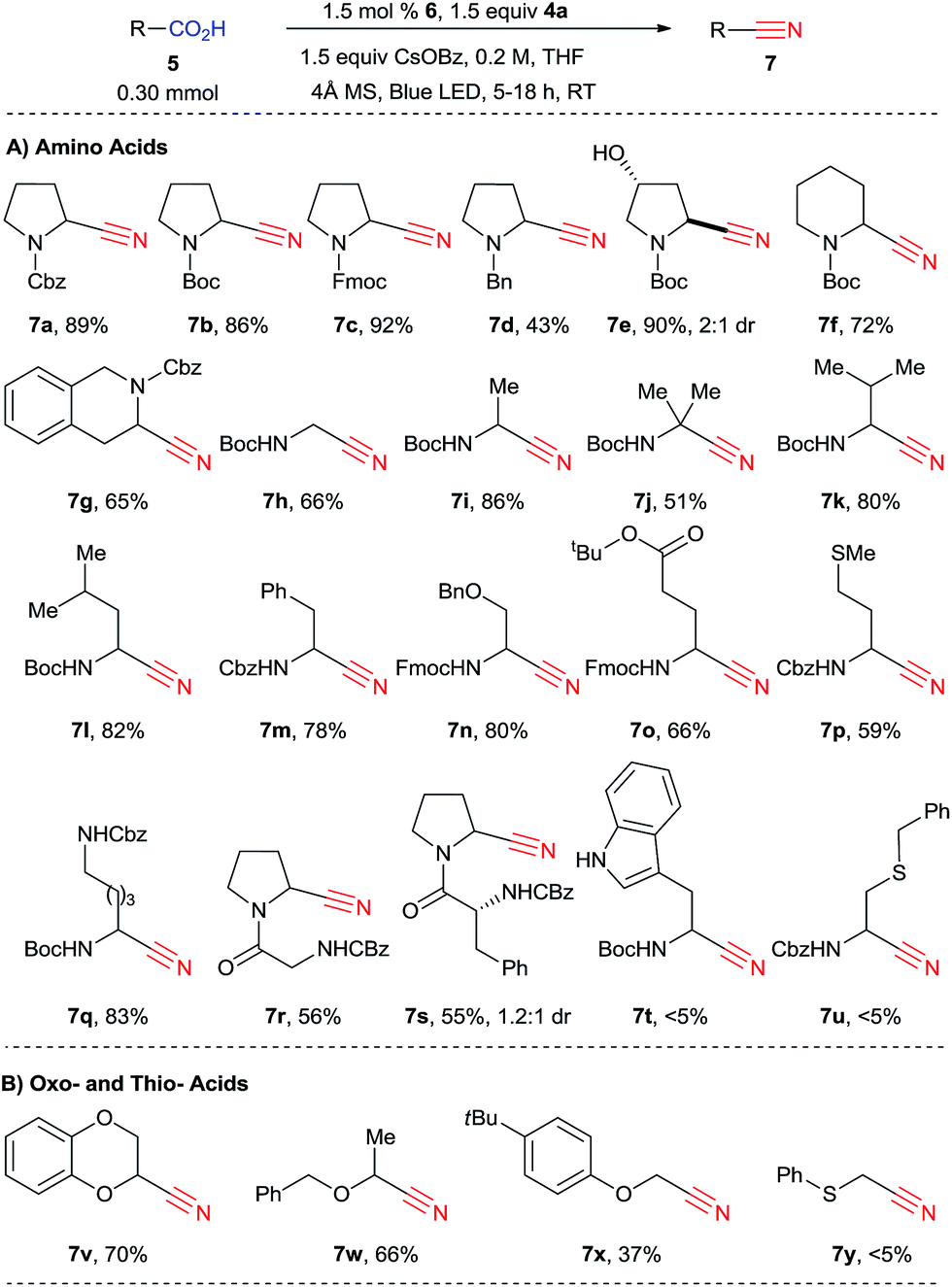 | ||
| Scheme 4 Scope of carboxylic acids. Reaction conditions: carboxylic acid (5, 0.30 mmol, 1.0 equiv.), CBX reagent (4a, 0.45 mmol, 1.5 equiv.), 6 (4.5 μmol, 0.015 equiv.), CsOBz (0.45 mmol, 1.5 equiv.), 4 Å molecular sieves (30 mg), THF (1.5 mL), 25–34 °C, blue LEDs irradiation for 5 to 18 h. Isolated yield after purification by column chromatography is given. | ||
We then turned to other classes of substrates and were pleased to see that oxy-acids also underwent decarboxylative cyanation. Cyclic or acyclic compounds are both suitable for the reaction (products 7v–x). Lower yield was obtained with an acyclic phenol ether (product 7x). α-Thio cyanide 7y could not be obtained under these reaction conditions. Furthermore, in contrast to the alkynylation reaction, only low yields were obtained in the case of carboxylic acids lacking the α heteroatom (<20%, results not shown). In this case, the major product obtained were the anhydrides resulting from the condensation of two carboxylic acids 5 or one carboxylic acid 5 and benzoic acid.
We then wondered if natural light could be used to promote the reaction. Indeed, after only four hours of sunlight irradiation, 7a was obtained in 90% yield (compared with 89% for blue LEDs, Scheme 5A). The reaction can also be scaled up to 1 mmol using only 0.1 mol% of catalyst 6, with a slight decrease of yield, as 7a was obtained in 60% yield after 48 h of irradiation (corresponding to 600 turnovers, Scheme 5B). To further highlight the utility of our methodology, 1,4-benzodioxan-2-carbonitrile (7v) was synthesized at the gram scale in 44% yield from the corresponding acid 5v (Scheme 5C). The drop in yield is probably due to the less efficient irradiation on larger scale. Nitrile 7v is the common key intermediate in the synthesis of various types of receptor antagonists (calcium, imidazoline, α2-adrenoreceptor), such as commercialized idazoxan (10) or lead compound WB-4101 (11) (Scheme 5).27 Another interesting application is the cyanation of carboxylic acid 5z, which can be obtained in one step from proline. Building block 7z can then be used to access the important antidiabetic drug vildagliptin (12) in only one step.28 However, acid 5z contains a highly reactive α-chloro amide unit, which was unfortunately not compatible with our standard reaction conditions. We speculated that cesium benzoate was reacting with the substrate due to its high nucleophilicity.29 Indeed, when potassium benzoate was used as base, the desired product 7z could be obtained in 42% yield.
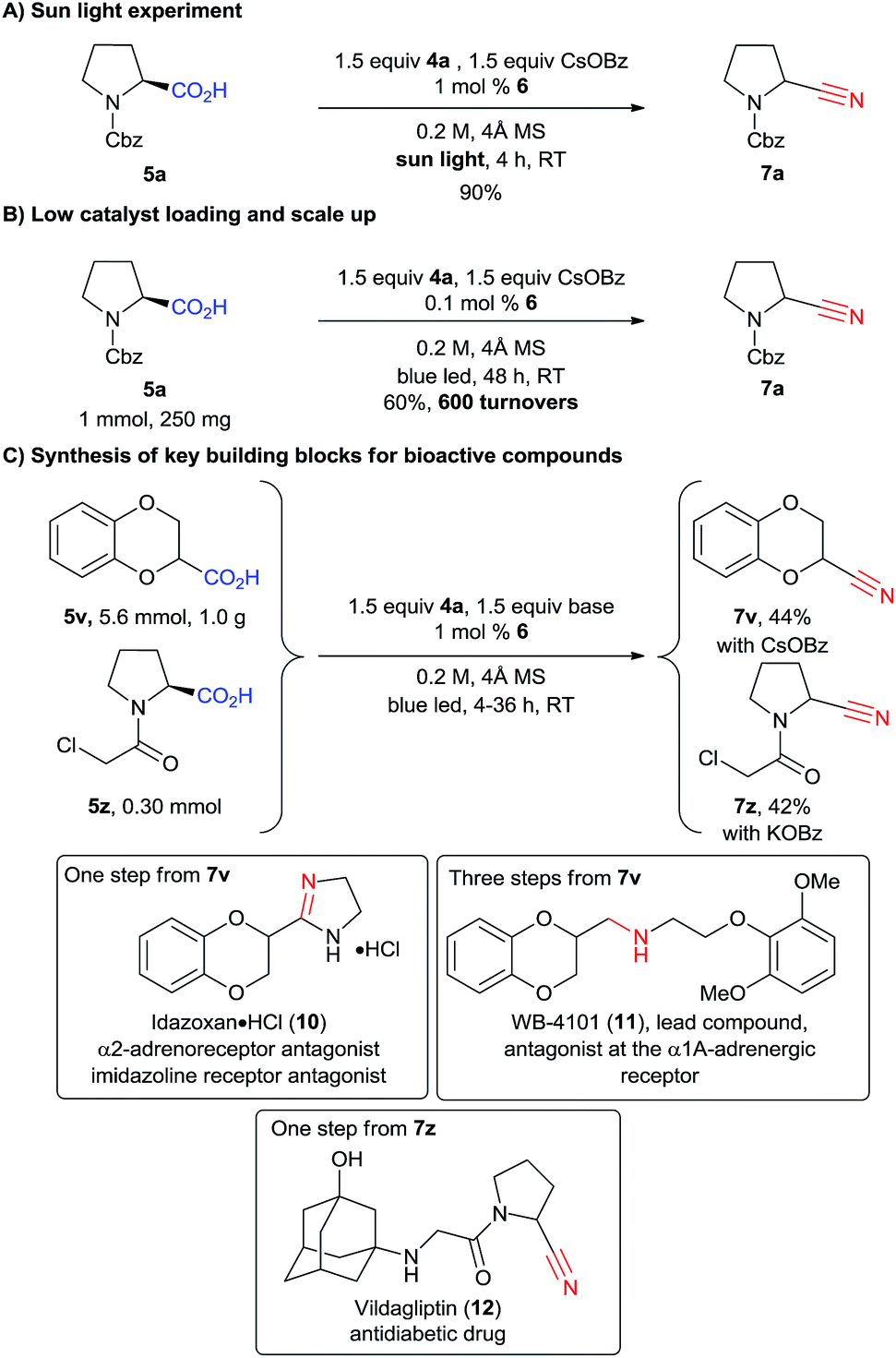 | ||
| Scheme 5 Sun light experiment (A), scale up (B) and synthesis of key building blocks (C). | ||
Mechanism and computational studies
When comparing the results obtained in our previous work on alkynylation20a with the decarboxylative cyanation, the transformations appear very similar upon first look: both reactions proceeded with the same catalyst and the benziodoxolone core of the hypervalent iodine reagents used was identical. Nevertheless, two important observations indicated that the reaction mechanism may be different:(1) The scope of the reaction was different: the alkynylation works with all classes of carboxylic acids. The presence of an α heteroatom is beneficial, but not crucial for success. On the other hand, the cyanation reaction had a broader scope than the alkynylation in the case of amino acids, but did not work well for simple aliphatic acids.
(2) Side product 8 observed in the presence of moisture for the cyanation reaction was not observed in the case of the alkynylation reaction.
Taken together, these results seemed to indicate that the cyanation reaction may occur via an intermediate with higher carbocation character. To support this speculation, the reaction mechanism was studied in more detail, both experimentally and via computation.
A speculative mechanism including different possible pathways is presented in Scheme 6.30 An important feature of this photoredox mediated Csp3–Csp coupling is the ability of carboxylic acids to undergo CO2 extrusion (Scheme 6A). It is now well established that Ir[dF(CF3)ppy]2(dtbbpy)PF6 (6) can generate the excited state *Ir[dF(CF3)ppy]2(dtbbpy)PF6 (6*) under visible light irradiation.15 This catalytic species is strongly oxidizing (E1/2(Ir*III/IrII) = +1.21 V vs. SCE) and can lead to a thermodynamically favored single electron transfer (SET) with the in situ generated cesium carboxylate I (+0.95 V for Boc-Pro-OCs vs. SCE),19f generating the strongly reducing Ir(II) complex 6red and the carboxy radical which undergoes immediate decarboxylation to give nucleophilic radical II. Intermediate II can then react with the hypervalent iodine reagents to give iodine centered radical III. To close the catalytic cycle, we assume that radical III can be reduced by the strongly reducing Ir(II) 6red (E1/2(IrIII/IrII) = −1.37 V vs. SCE), thus regenerating the ground state photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (6).
 | ||
| Scheme 6 Speculative mechanism for the decarboxylative cyanation and alkynylation reactions. | ||
Both the alkynylation and cyanation products were racemic, supporting the formation of either a radical or carbocation intermediate. To further support the existence of carbon centered radical II, we turned to radical clock and/or trapping experiments (Scheme 7). A radical clock experiment with cyclopropane 16 and EBX reagent 13b led to the formation of ring-opening product 17, confirming the intermediacy of radicals in the case of the alkynylation reaction. A similar experiment has also been done by Xiao and co-workers.20b In order to have a radical clock which could be used in both reactions, we then examined cyclopropyl amino acid 18. In the case of the alkynylation reaction, alkyne 19 could be isolated in 20% yield. This product probably resulted from the hydrolysis of the expected enamide 20. However, we were not able to isolate any product from the corresponding cyanation reaction. We therefore attempted a radical trap experiment with TEMPO in the cyanation reaction of protected proline 5a. In this case, the formation of the cyanation product was completely inhibited, and a mass corresponding to TEMPO-adduct 21 could be observed by high resolution mass spectrometry. The presence of radical intermediate II is therefore strongly supported for the alkynylation reaction. For the cyanation, it can be only considered as probable at this stage, as TEMPO can also act as a SET reagents and not only as a radical trap when photoredox catalytic cycles are considered.
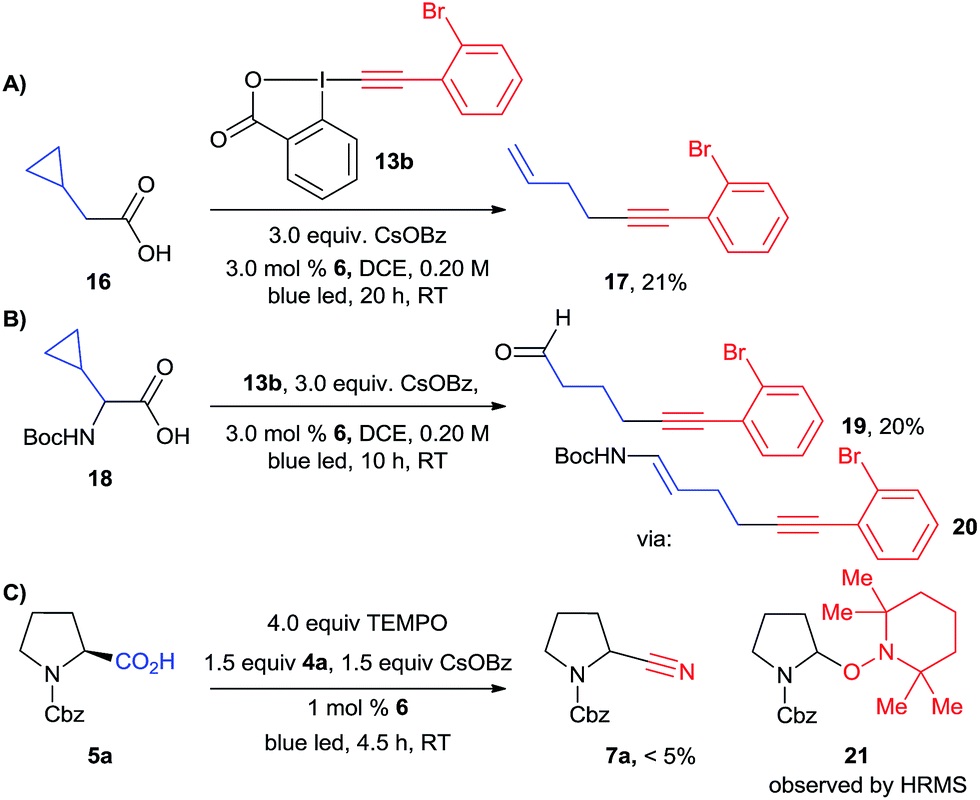 | ||
| Scheme 7 Experiments supporting the existence of a carbon centered radical intermediate II. | ||
After alkynylation or cyanation, the catalytic cycle would be closed by reduction of the formed radical III by iridium complex 6red.31 It is very challenging to gain further information about this catalytic step, due to the high reactivity of intermediate III. Nevertheless, recent computations performed by Chen and co-workers supported the fact that radical III is best described as a resonance structure including an iodine and an oxygen centered radical.32 The resulting enhanced stability may have several effects: first, it will make formation of the radical easier, and therefore accelerate the cyanation or alkynylation step. Second, it should make reduction more difficult, rationalizing the need for a photoredox catalyst with a relatively strong reduction potential. To support this hypothesis, we computed the reduction potential of radical III, as this value could not be obtained so far experimentally, and obtained 0.25 V.33 The reduction potential of complex 6red is −1.37 V vs. SCE, therefore the speculated catalytic step at least appears thermodynamically feasible. Third, decarboxylation to give an aryl radical becomes more difficult, preventing potential side reactions resulting from these highly reactive species.
With a crude picture of the general mechanism in hand, we then turned to the investigation of the key alkynylation/cyanation step (Scheme 6B). Li and co-workers proposed α-addition followed by β-elimination as mechanism for the reaction of radicals with EBX reagents in their seminal work in 2012 (path a).34 This mechanism could also be proposed for the cyanation reaction. However, based on our work on the reaction of thiol anions and radicals with EBX and CBX reagents,22b,35 which highlighted a more complex mechanistic picture, we wondered if other reaction pathways would also be accessible for carbon centered radicals. In particular, a one-step concerted α-addition/elimination mechanism could also be considered (path b). Furthermore, it is difficult to exclude directly a mechanism involving β-addition, followed by α-elimination and 1,2-shift (path c). Nevertheless, this mechanism appears less probable in the case of the cyanation reaction, as stable isonitrile products should have been isolated. However, none of these mechanisms would explain well the differences observed between the cyanation and the alkynylation reactions. In particular, the formation of alcohol 8 in the presence of water and absence of oxygen strongly supports the existence of an iminium intermediate in the case of proline derivative 5a. Indeed, when the reaction was run in presence of 18O labelled water, isotope incorporation in product 8 was observed. We therefore propose a new reaction mechanism involving single electron transfer from radical II to the benziodoxolone reagent to give the radical ion pair d1 (path d). In fact, the oxidation of α-amino radicals to iminiums is well established.36 Collapse of the radical anion to give the carbanion followed by recombination with the carbocation will lead to the observed product and generate iodine centered radical III. In principle, activated iridium catalyst 6* could also oxidize radical II, but as both species are present in catalytic amounts, this appears less probable. In this case, cyanation should also be observed in the presence of a nucleophilic cyanation reagent, but no product was obtained when the reaction was done in the presence of KCN without CBX (4a). Furthermore, formation of alcohol 8 would have been expected independently of the reagent used, and it was observed only in the case of cyanation. Our working hypothesis was, therefore, that path d would be favored for CBX reagents, but not for EBX.
To gain further insight into the reactivity differences between the two classes of reagents, competition experiments were run between TIPS-EBX (13a) and CBX (4a) on proline derivative 5a. Cyanation was favored, showing the higher reactivity of CBX (4a). This result allowed us to exclude that the formation of side product 8 was avoided by a faster reaction in the case of the alkynylation reaction. To further support the presence of an iminium intermediate, we ran the cyanation reaction in the presence of C13 labelled potassium cyanide. Indeed, 2.2% C13 incorporation was observed. However, a control experiment showed that cyanide exchange was occurring directly on CBX (4a) under the reaction conditions. Consequently, this experiment cannot be used to further support the existence of an iminium intermediate.
Therefore, we turned to density functional theory (DFT) computations to further support two different mechanistic pathways (Fig. 2). Both the alkynylation and the cyanation of proline derivatives 5a with TIPS-EBX (13a) and CBX (4a) were computed at the PBE0-dDsC/TZ2P//M06/def2-SVP theoretical level (see Computational details for additional information) for mechanistic paths b–d (Fig. 2). In order to reproduce the solvent effect, an implicit continuum model for realistic solvents (COSMO-RS) was used, with DCE for the alkynylation and THF for the cyanation. For both reactions, we were unable to locate a reaction intermediate corresponding to the frequently proposed radical intermediate a1 following path a. Therefore, only paths b–d are represented. For the alkynylation reaction, both path b and c starts with a van der Waals interaction complex b0/c0, the formation of which is endothermic (Fig. 2A). From this intermediate, both a transition state bTS1 leading directly to the alkynylation product 14avia concerted α addition and a transition state cTS1 leading to radical intermediate c1via β-addition could be located. The energies of both transition states are very close, indicating that the reaction could follow both pathways simultaneously. From radical c1, bond dissociation to generate radical III is followed by a barrierless 1,2-silicon shift to give alkynylation product 14a. Finally, the SET pathway d was computed. For the steps involving electron-transfer, no energy barrier was determined for the outer sphere transfer mechanism.37 Electron-transfer from TIPS-EBX (13a) to radical II was found to be feasible, with only 11.3 kcal mol−1 required. However, the collapse of the radical anion to form radical III and an acetylide anion was found to be highly unfavorable, with an energy of 32.8 kcal mol−1. Consequently, even if the SET transfer occur between TIPS-EBX (13a) and radical II, it is probably not contributing to the alkynylation reaction.
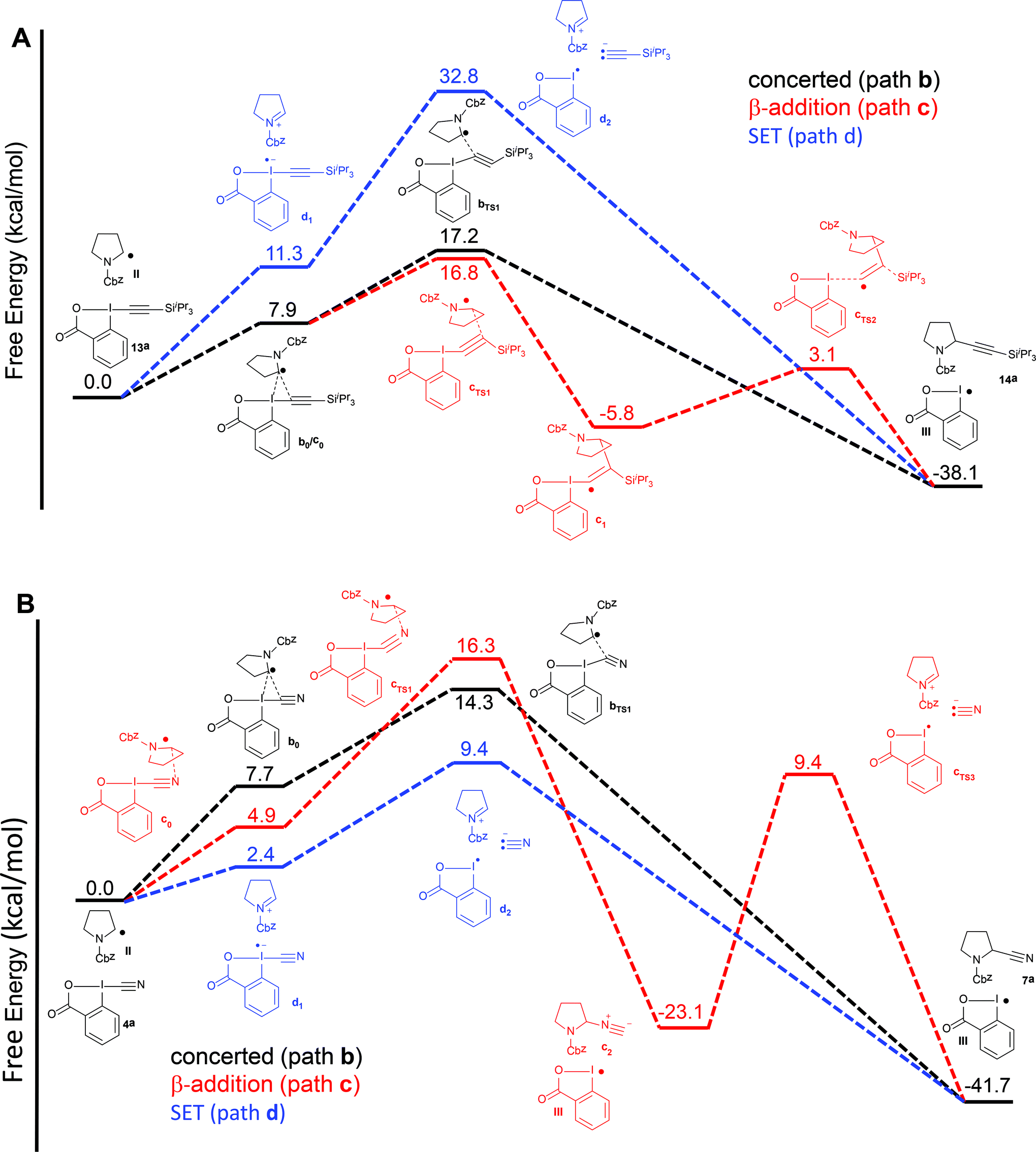 | ||
| Fig. 2 Reaction free energy profile at the [PBE0-dDsC/TZ2P//M06/def2-SVP level for the alkynylation (A) and cyanation (B) of protected proline 5a for paths b–d. | ||
Pathways b–d were then examined for the cyanation reaction (Fig. 2B). α-Addition via interaction complex b0 and transition state bTS1 occurred relatively easily, with a slightly lower transition state energy than the related alkynylation reaction (14.3 vs. 17.2 kcal mol−1). In contrast to the alkynylation reaction however, the β addition pathways was higher in energy (16.3 kcal mol−1), in accordance with the fact that no isonitrile product had been observed. In this case, intermediate c1 could not be located, and formation of isonitrile c2 was directly observed. As expected, conversion of isonitrile c2 to the cyanation product 7a was predicted to be difficult, as the lowest energy pathway involved heterolytic bond cleavage with an activation energy of 34.5 kcal mol−1. However, the major difference in the cyanation reaction appeared when the SET pathway d was computed. First, the oxidation of radical II by CBX (4a) is much easier than with TIPS-EBX (13a) (2.4 vs. 11.3 kcal mol−1). Second and most importantly, the collapse of the formed radical anion is again easy, as in this case the formation of the cyanide anion and radical III requires only 9.4 kcal mol−1. Even if the assumption of a barrierless electron transfer could lead to an underestimation of the activation energy for this process, it appears plausible that the cyanation reaction could occur via a SET pathway, whereas this looks highly improbable for the alkynylation reaction.
The easier electron transfer to CBX (4a) when compared to TIPS-EBX (13a) could indicate a higher reduction potential. Both reagents were therefore examined by cyclic voltammetry. Although no defined reduction wave could be identified in the case of TIPS-EBX (13a), CBX (4a) showed an irreversible system with a clear reduction wave at −0.92 V vs. SCE.38 This confirms that CBX is a relatively strong oxidant, which should be able to oxidize α-amino radicals to the corresponding iminium.
3. Conclusion
In summary, we have developed a one-step decarboxylative cyanation of α-amino and α-oxy acids using cyanobenziodoxolone (CBX, 4a). The reaction proceeded at room temperature under visible light irradiation using 0.1–1.5 mol% of an iridium catalyst. In particular, a broad range of amino acids could be cyanated using this methodology. Combined experimental and computational studies indicated that the favored mechanism is probably different from the previously developed decarboxylative alkynylation. Direct reaction of the radical formed by iridium-mediated decarboxylation was lower in energy for the alkynylation, whereas single electron transfer (SET) to form an iminium intermediate followed by cyanide addition was favored for cyanation. The cyanation reaction is expected to have high value for the synthesis of useful nitrile building blocks from biomass, whereas the discovery of different mechanistic pathways for the reaction of radicals with benziodoxole reagents will set the bases for the development of further transformations based on the use of these versatile compounds.4. Computational details
Geometries of minima and transition states were optimized using the M0639 density functional with the def2-SVP basis set in Gaussian 09.40 M06 computations uniformly employed the “Ultrafine” grid to remove known problems with integration grid size.41 Refined energy estimates that explicitly account for non-bonded interactions were obtained using a density-dependent dispersion correction42 appended to the PBE043 functional (PBE0-dDsC). PBE0-dDsC single point computations used the TZ2P, as implemented in ADF.44 All free energies include the effects of solvation using the implicit continuum model for realistic solvents45 (COSMO-RS), as implemented in ADF, as well as unscaled free energy corrections derived from M06/def2-SVP computations. Reported reduction potentials were determined at the M06/def2-TZVPP level.Acknowledgements
We thank EPFL and European Research Council for financial support (ERC; Starting Grant iTools4MC, number 334840). The Laboratory for Computational Molecular Design at EPFL is acknowledged for providing computational resources. F. L. V. thanks Dr Stefano Nicolai and Dr Durga P. Hari from our laboratory for helpful suggestions and comments. Dr Thomas Di Franco from the Laboratory of Inorganic Synthesis and Catalysis at EPFL is acknowledged for his help in the measurement of cyclic voltammograms. Xavier Jeanbourquin from Laboratory of Molecular Engineering of Optoelectronics Nanomaterials at EPFL is acknowledged for his help in the measurement of quantum yields. Dr Ruud Hovius from the Laboratory of Protein Engineering at EPFL is thanked for his help in performing the Stern–Volmer experiment.Notes and references
- (a) Z. Rappoport, Chemistry of the Cyano Group, Wiley, London, 1970 Search PubMed; (b) F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597 RSC; (c) D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359 RSC; (d) F. F. Fleming and Q. Z. Wang, Chem. Rev., 2003, 103, 2035 CrossRef CAS PubMed; (e) S. T. Murphy, H. L. Case, E. Ellsworth, S. Hagen, M. Huband, T. Joannides, C. Limberakis, K. R. Marotti, A. M. Ottolini, M. Rauckhorst, J. Starr, M. Stier, C. Taylor, T. Zhu, A. Blaser, W. A. Denny, G.-L. Lu, J. B. Smaill and F. Rivault, Bioorg. Med. Chem. Lett., 2007, 17, 2150 CrossRef CAS PubMed; (f) J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed; (g) T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137 CrossRef CAS PubMed; (h) Y. Ping, Q. Ding and Y. Peng, ACS Catal., 2016, 6, 5989 CrossRef CAS.
- (a) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed; (b) M. A. Perry, M. D. Morin, B. W. Slafer and S. D. Rychnovsky, J. Org. Chem., 2012, 77, 3390 CrossRef CAS PubMed.
- (a) J. M. Nabholtz, A. Buzdar, M. Pollak, W. Harwin, G. Burton, A. Mangalik, M. Steinberg, A. Webster and M. von Euler, J. Clin. Oncol., 2000, 18, 3758 CAS; (b) D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, A. Khanna, J. G. Robertson, A. Y. Wang, L. M. Simpkins, P. Taunk, Q. Huang, S. P. Han, B. Abboa-Offei, M. Cap, L. Xin, L. Tao, E. Tozzo, G. E. Welzel, D. M. Egan, J. Marcinkeviciene, S. Y. Chang, S. A. Biller, M. S. Kirby, R. A. Parker and L. G. Hamann, J. Med. Chem., 2005, 48, 5025 CrossRef CAS PubMed; (c) J. Y. Gauthier, N. Chauret, W. Cromlish, S. Desmarais, L. T. Duong, J. P. Falgueyret, D. B. Kimmel, S. Lamontagne, S. Leger, T. LeRiche, C. S. Li, F. Masse, D. J. McKay, D. A. Nicoll-Griffith, R. A. Oballa, J. T. Palmer, M. D. Percival, D. Riendeau, J. Robichaud, G. A. Rodan, S. B. Rodan, C. Seto, M. Therien, V. L. Truong, M. C. Venuti, G. Wesolowski, R. N. Young, R. Zamboni and W. C. Black, Bioorg. Med. Chem. Lett., 2008, 18, 923 CrossRef CAS PubMed.
- (a) Z. P. Demko and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2110 CrossRef CAS; (b) Z. P. Demko and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2113 CrossRef CAS.
- P. Lassalas, B. Gay, C. Lasfargeas, M. J. James, V. Tran, K. G. Vijayendran, K. R. Brunden, M. C. Kozlowski, C. J. Thomas, A. B. Smith, D. M. Huryn and C. Ballatore, J. Med. Chem., 2016, 59, 3183 CrossRef CAS PubMed.
- D. T. Mowry, Chem. Rev., 1948, 42, 189 CrossRef CAS PubMed.
- Selected examples: (a) T.-J. Gong, B. Xiao, W.-M. Cheng, W. Su, J. Xu, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 10630 CrossRef CAS PubMed; (b) Z. B. Shu, W. Z. Ji, X. Wang, Y. J. Zhou, Y. Zhang and J. B. Wang, Angew. Chem., Int. Ed., 2014, 53, 2186 CrossRef CAS PubMed; (c) Y. Yang and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 8677 CrossRef CAS PubMed; (d) D. G. Yu, T. Gensch, F. de Azambuja, S. Vasquez-Cespedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS PubMed; (e) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635 CrossRef CAS PubMed; (f) M. Chaitanya and P. Anbarasan, Org. Lett., 2015, 17, 3766 CrossRef CAS PubMed.
- Selected examples: (a) Z. M. Zheng and C. L. Hill, Chem. Commun., 1998, 2467 RSC; (b) S. I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931 CrossRef CAS PubMed; (c) S. I. Murahashi, T. Nakae, H. Terai and N. Komiya, J. Am. Chem. Soc., 2008, 130, 11005 CrossRef CAS PubMed; (d) S. Singhal, S. L. Jain and B. Sain, Chem. Commun., 2009, 2371 RSC; (e) W. Han and A. R. Ofial, Chem. Commun., 2009, 5024 RSC; (f) A. Wagner, W. Han, P. Mayer and A. R. Ofial, Adv. Synth. Catal., 2013, 355, 3058 CrossRef CAS; (g) A. Wagner, N. Hampel, H. Zipse and A. R. Ofial, Org. Lett., 2015, 17, 4770 CrossRef CAS PubMed; (h) Y. Zhang, H. Peng, M. Zhang, Y. Cheng and C. Zhu, Chem. Commun., 2011, 47, 2354 RSC; (i) N. Sakai, A. Mutsuro, R. Ikeda and T. Konakahara, Synlett, 2013, 24, 1283 CrossRef CAS.
- Selected examples: (a) M. North, Angew. Chem., Int. Ed., 2004, 43, 4126 CrossRef CAS PubMed; (b) T. Tajima and A. Nakajima, J. Am. Chem. Soc., 2008, 130, 10496 CrossRef CAS PubMed; (c) S. Kamijo, T. Hoshikawa and M. Inoue, Org. Lett., 2011, 13, 5928 CrossRef CAS PubMed; (d) L. Ma, W. Chen and D. Seidel, J. Am. Chem. Soc., 2012, 134, 15305 CrossRef CAS PubMed; (e) T. Hoshikawa, S. Yoshioka, S. Kamijo and M. Inoue, Synthesis, 2013, 45, 874 CrossRef CAS; (f) D. B. Ushakov, K. Gilmore, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 53, 557 CrossRef CAS PubMed; (g) S. S. Kong, L. Q. Zhang, X. L. Dai, L. Z. Tao, C. S. Xie, L. Shi and M. Wang, Adv. Synth. Catal., 2015, 357, 2453 CrossRef CAS; (h) A. M. Nauth, N. Otto and T. Opatz, Adv. Synth. Catal., 2015, 357, 3424 CrossRef CAS; (i) H. Shen, X. Zhang, Q. Liu, J. Pan, W. Hu, Y. Xiong and X. Zhu, Tetrahedron Lett., 2015, 56, 5628 CrossRef CAS.
- (a) B. A. Halkier, H. V. Scheller and B. L. Moller, in Cyanide Compounds in Biology (Ciba Foundation Symposium 140), Wiley, Chichester, 1988, p. 49 Search PubMed; (b) M. T. Nelp and V. Bandarian, Angew. Chem., Int. Ed., 2015, 54, 10627 CrossRef CAS PubMed.
- (a) R. Graf, Chem. Ber., 1956, 89, 1071 CrossRef CAS; (b) G. Lohaus, Chem. Ber., 1967, 100, 2719 CrossRef CAS; (c) K. Miyagi, K. Moriyama and H. Togo, Eur. J. Org. Chem., 2013, 5886 CrossRef CAS.
- (a) D. A. Klein, J. Org. Chem., 1971, 36, 3050 CrossRef CAS; (b) D. Cantillo and C. O. Kappe, J. Org. Chem., 2013, 78, 10567 CrossRef CAS PubMed; (c) D. Cartigny, A. Dos Santos, L. El Kaim, L. Grimaud, R. Jacquot and P. Marion, Synthesis, 2014, 46, 1802 CrossRef.
- (a) D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939 RSC; (b) D. H. R. Barton, J. C. Jaszberenyi and E. A. Theodorakis, Tetrahedron, 1992, 48, 2613 CrossRef CAS.
- (a) S. Kim and C. J. Lim, Angew. Chem., Int. Ed., 2002, 41, 3265 CrossRef CAS; (b) A.-P. Schaffner, V. Darmency and P. Renaud, Angew. Chem., Int. Ed., 2006, 45, 5847 CrossRef CAS PubMed; (c) B. Gaspar and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 4519 CrossRef CAS PubMed; (d) C. H. Cho, J. Y. Lee and S. Kim, Synlett, 2009, 81 CAS.
- Selected examples and reviews: (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed; (e) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (g) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (h) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed.
- M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 12709 RSC.
- J. C. O. Pacheco, A. Lipp, A. M. Nauth, F. Acke, J. P. Dietz and T. Opatz, Chem.–Eur. J., 2016, 22, 5409 CrossRef PubMed.
- J. J. Dai, W. M. Zhang, Y. J. Shu, Y. Y. Sun, J. Xu, Y. S. Feng and H. J. Xu, Chem. Commun., 2016, 52, 6793 RSC.
- Selected examples: (a) L. Chen, C. S. Chao, Y. Pan, S. Dong, Y. C. Teo, J. Wang and C.-H. Tan, Org. Biomol. Chem., 2013, 11, 5922 RSC; (b) Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 7854 RSC; (c) J. Xie, P. Xu, H. Li, Q. Xue, H. Jin, Y. Cheng and C. Zhu, Chem. Commun., 2013, 49, 5672 RSC; (d) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (e) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886 CrossRef CAS PubMed; (f) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS PubMed; (g) A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624 CrossRef CAS PubMed; (h) S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654 CrossRef CAS PubMed; (i) J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 502 CrossRef CAS PubMed; (j) M. Rueda-Becerril, O. Mahe, M. Drouin, M. B. Majewski, J. G. West, M. O. Wolf, G. M. Sammis and J.-F. Paquin, J. Am. Chem. Soc., 2014, 136, 2637 CrossRef CAS PubMed; (k) C. Cassani, G. Bergonzini and C.-J. Wallentin, Org. Lett., 2014, 16, 4228 CrossRef CAS PubMed; (l) P. Xu, A. Abdukader, K. Hu, Y. Cheng and C. Zhu, Chem. Commun., 2014, 50, 2308 RSC; Review: (m) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef CAS PubMed.
- (a) F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200 CrossRef CAS PubMed; (b) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196 CrossRef CAS PubMed.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz, B. Mismash, J. K. Woodward and A. J. Simonsen, Tetrahedron Lett., 1995, 36, 7975 CrossRef CAS.
- (a) Y. F. Wang, J. S. Qiu, D. J. Kong, Y. T. Gao, F. P. Lu, P. G. Karmaker and F. X. Chen, Org. Biomol. Chem., 2015, 13, 365 RSC; (b) R. Frei, T. Courant, M. D. Wodrich and J. Waser, Chem.–Eur. J., 2015, 21, 2662 CrossRef CAS PubMed; (c) R. Chowdhury, J. Schorgenhumer, J. Novacek and M. Waser, Tetrahedron Lett., 2015, 56, 1911 CrossRef CAS PubMed; (d) M. Chen, Z. T. Huang and Q. Y. Zheng, Org. Biomol. Chem., 2015, 13, 8812 RSC; (e) M. V. Vita, P. Caramenti and J. Waser, Org. Lett., 2015, 17, 5832 CrossRef CAS PubMed; For recent review on hypervalent iodine reagents, see: (f) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436 CrossRef CAS PubMed; (g) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed.
- During preparation of this manuscript, Xiao and co-workers reported the cyanation of tertiary amines using cyanobenziodoxolones under photoredox conditions. In this work, two examples of decarboxylative cyanation in moderate yields were also reported. However, the authors proposed a radical-based mechanism for this transformation: Q. Q. Zhou, D. Liu, W. J. Xiao and L. Q. Liu, Acta Chim. Sin., 2016 DOI:10.6023/a16080414 , in press.
- When cheaper organic dyes such as DCA (9,10-anthracenedicarbonitrile) and 9-mesityl-10-methylacridinium perchlorate were used, no product was obtained under the reaction conditions.
- Determined by integration of the 1H NMR signals in the crude mixture.
- (a) W. A. Schroeder, J. B. Shelton and J. R. Shelton, Arch. Biochem. Biophys., 1969, 130, 551 CrossRef CAS PubMed; (b) A. S. Inglis and P. Edman, Anal. Biochem., 1970, 37, 73 CrossRef CAS PubMed.
- (a) C. B. Chapleo, P. L. Myers, R. C. M. Butler, J. C. Doxey, A. G. Roach and C. F. C. Smith, J. Med. Chem., 1983, 26, 823 CrossRef CAS PubMed; (b) D. Giardina, P. Angeli, L. Brasili, U. Gulini, C. Melchiorre and G. Strappaghetti, Eur. J. Med. Chem., 1984, 19, 411 CAS.
- L. Pellegatti and J. Sedelmeier, Org. Process Res. Dev., 2015, 19, 551 CrossRef CAS.
- G. Dijkstra, W. H. Kruizinga and R. M. Kellogg, J. Org. Chem., 1987, 52, 4230 CrossRef CAS.
- Alternatively reduction of CBX (4a) by the iridium catalyst 6red to give radical III and cyanide could also be considered. To complete the catalytic cycle, radical III could recombine with the α-amino radical II. The formed unstable hemiaminal could then give the iminium, which would react with the cyanide anion. Although this catalytic cycle cannot be excluded at this stage, it appears less probable, as the formation of the hemi-acetal derived from 2-iodobenzoic acid was not observed during the reaction, and computation shows that radical II can react very fast with CBX (4a), present in stoichiometric amount.
- In principle, a radical chain could also be initiated under the reaction conditions. However, the quantum yield of the reaction was determined to be 79% and 88% for alkynylation and cyanation respectively. In case of a chain reaction, quantum yield higher than 100% are more frequently observed. Furthermore, a catalytic cycle starting with electron-transfer from CBX (4a) to activated catalyst 6* was excluded by a Stern–Volmer analysis: excited state quenching was observed with carboxylate I, but not with CBX (4a). See ESI† for further details.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407 RSC.
- A value of 0.66 V was obtained for a simple benzoyl radical. This higher oxidation potential is in agreement with the stabilization of radical IIIvia resonance.
- X. Liu, Z. Wang, X. Cheng and C. Li, J. Am. Chem. Soc., 2012, 134, 14330 CrossRef CAS PubMed.
- (a) R. Frei and J. Waser, J. Am. Chem. Soc., 2013, 135, 9620 CrossRef CAS PubMed; (b) R. Frei, M. D. Wodrich, D. P. Hari, P. A. Borin, C. Chauvier and J. Waser, J. Am. Chem. Soc., 2014, 136, 16563 CrossRef CAS PubMed; (c) M. D. Wodrich, P. Caramenti and J. Waser, Org. Lett., 2016, 18, 60 CrossRef CAS PubMed.
- (a) P. J. DeLaive, J. T. Lee, H. W. Sprintschnik, H. Abruna, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1977, 99, 7094 CrossRef CAS; (b) D. D. M. Wayner, J. J. Dannenberg and D. Griller, Chem. Phys. Lett., 1986, 131, 189 CrossRef CAS; (c) C. Dai, F. Meschini, J. M. R. Narayanam and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425 CrossRef CAS PubMed; (d) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977 CrossRef PubMed.
- In principle, a more accurate reproduction of the free energy associated with outer sphere electron transfer could be determined, for example using molecular dynamics and explicit solvent to reproduce the reorganization energy. However, such computations are beyond the scope of the simplified mechanistic picture presented here.
- See Fig. S1 in the ESI.†.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 15 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. E. Wheeler and K. N. Houk, J. Chem. Theory Comput., 2010, 6, 395 CrossRef CAS PubMed.
- (a) S. N. Steinmann and C. Corminboeuf, J. Chem. Theory Comput., 2011, 7, 3567 CrossRef CAS PubMed; (b) S. N. Steinmann and C. Corminboeuf, J. Chem. Phys., 2011, 134, 044117 CrossRef PubMed; (c) S. N. Steinmann and C. Corminboeuf, Chimia, 2011, 65, 240 CrossRef CAS PubMed; (d) S. N. Steinmann and C. Corminboeuf, J. Chem. Theory Comput., 2010, 6, 1990 CrossRef CAS PubMed.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (b) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS; (b) C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391 Search PubMed.
- A. Klamt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 699 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04907a |
| This journal is © The Royal Society of Chemistry 2017 |