
Hydroamination of alkenyl N-arylhydrazones mediated by t-BuOK for the synthesis of nitrogen heterocycles†
Xingao
Peng
,
Atsushi
Kaga
,
Hajime
Hirao
* and
Shunsuke
Chiba
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: hirao@ntu.edu.sg; shunsuke@ntu.edu.sg
First published on 9th March 2016
Abstract
The t-BuOK-mediated reactions of γ,δ-alkenyl N-arylhydrazones enabled intramolecular hydroamination with the outer nitrogen, affording tetrahydropyridazine derivatives. DFT calculations demonstrated a clear distinction in the chemical reactivity between hydrazones and analogous oximes in inorganic base-mediated hydroamination.
 Shunsuke Chiba | Shunsuke Chiba earned his Ph.D. in 2006 from the University of Tokyo (Prof. Koichi Narasaka). He was appointed as a research associate at the University of Tokyo in 2005. He began his independent career at Nanyang Technological University (Singapore) as an Assistant Professor in 2007. In 2012, he was promoted to Associate Professor (with tenure) in the same university. His research focus is on methodology development in the area of synthetic organic chemistry. |
As one of the most efficient ways to construct nitrogen-heterocycles,1,2 intramolecular hydroamination of alkenyl amines and their derivatives has been intensively studied with various metal complexes (alkali metals, transition metals, and f-block elements) as well as Brønsted acids.3,4 We have recently studied inorganic base-mediated hydroamination of alkenyl oximes and hydrazones for the synthesis of nitrogen-heterocycles.5 The reactions of γ,δ-alkenyl oximes mediated by potassium bases such as K3PO4 and t-BuOK proceeded in an unprecedented fashion to yield 5-membered ring nitrones, which were formed via nucleophilic amination of inactivated alkenes by the oxime nitrogen (Scheme 1a).5a Density functional theory (DFT) calculations suggested that the ionic interaction between the potassium cation on the oxime oxygen and the negatively charged alkene moiety stabilizes the transition state. On the other hand, t-BuOK-mediated reactions of γ,δ-unsaturated N-alkylhydrazones led to the construction of pyrrolidine skeletons via hydrazone–hydrazone isomerization with a 1,3-proton shift followed by the nucleophilic hydroamination; in this reaction, modification of the substituents on the hydrazone moiety greatly affected the diastereoselectivity (Scheme 1b).5b Based on these findings, we wondered whether γ,δ-alkenyl N-arylhydrazones, for which hydrazone–hydrazone isomerization is not possible, undergo cyclization in the same way as the oximes, to form the corresponding 5-membered ring azomethineimines or afford tetrahydropyridazines via 6-exocyclization with the outer nitrogen.6,7 We herein report the results from our experimental and theoretical studies to address this question.
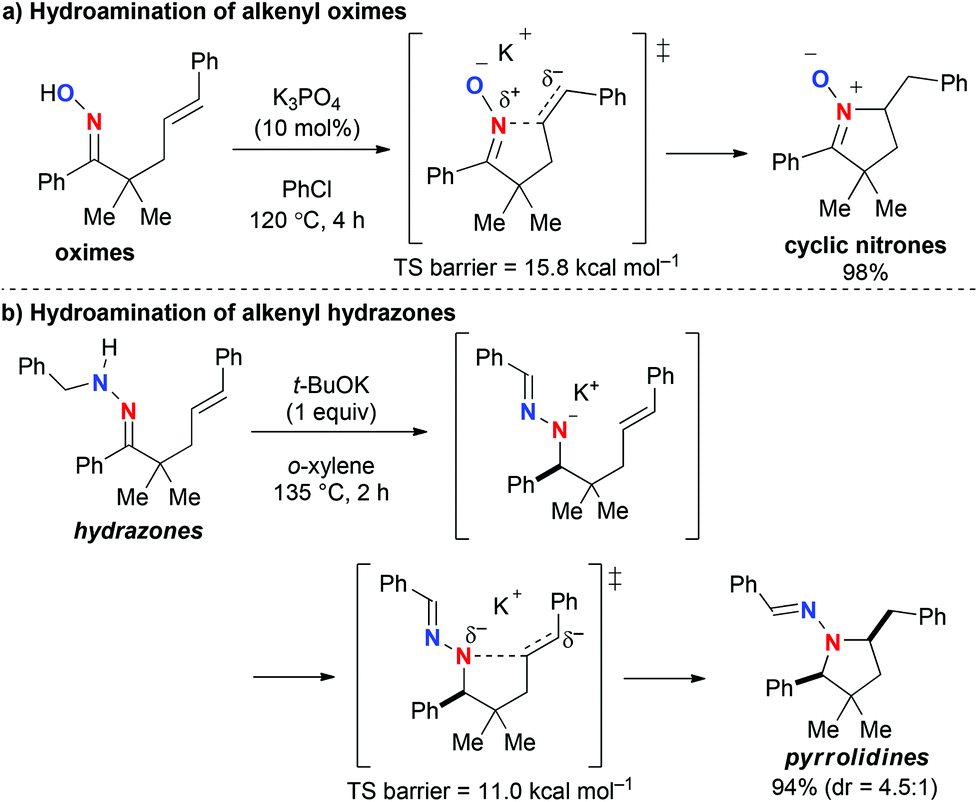 | ||
| Scheme 1 Inorganic base-mediated hydroamination of alkenyl oximes and hydrazones. | ||
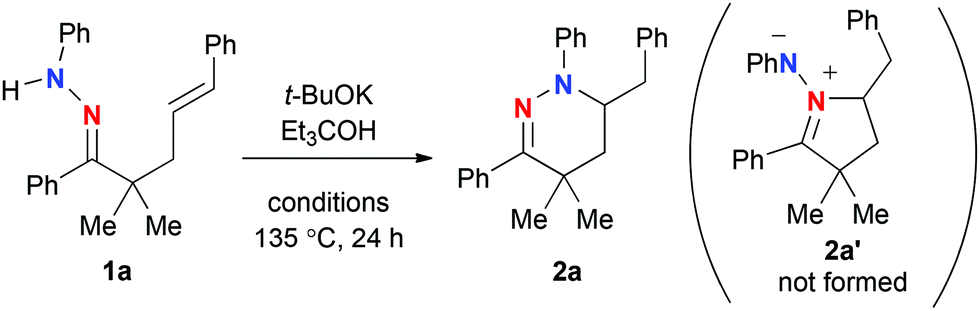
Our investigation was commenced with the reactions of γ,δ-alkenyl N-phenylhydrazone 1a (Table 1). The reaction with 1 equiv. of t-BuOK8 in o-xylene proceeded at 135 °C (entry 1), resulting in the formation of 6-membered ring tetrahydropyridazine 2a in 62% yield via hydroamination of the alkene with the outer nitrogen atom (marked in blue), while the formation of azomethineimine 2a′ was not observed at all. Further optimization of the reaction conditions revealed that addition of Et3COH as an additive could improve the yield of 2a (entry 2). The reactions with catalytic amounts of t-BuOK were not optimal for this reaction (entries 3 and 4). The reaction in DMF performed well (entry 5), while that in DMSO became sluggish (entry 6). It is noted that the reactions of 1a with t-BuOLi and t-BuONa as well as other potassium bases such as K3PO4 and K2CO3 gave no hydroamination product 2a.
|
|
||||
|---|---|---|---|---|
| Entry | t-BuOK (equiv.) | Et3COH (equiv.) | Solvent | Yield of 2ab (%) |
| a Unless otherwise noted, the reactions were carried out on the scale of 0.5 mmol of hydrazone 1a in the solvent (5 mL) under an Ar atmosphere. b Isolated yields were recorded. c Recovery yields of 1a based on 1H NMR. | ||||
| 1 | 1 | 0 | o-Xylene | 62 (23)c |
| 2 | 1 | 3 | o-Xylene | 84 |
| 3 | 0.2 | 3 | o-Xylene | 10 (82)c |
| 4 | 0.4 | 3 | o-Xylene | 44 (50)c |
| 5 | 1 | 3 | DMF | 62(11)c |
| 6 | 1 | 3 | DMSO | 42 |
Having observed the distinct reaction outcomes for inorganic-base mediated hydroamination of oximes (Scheme 1a) and N-phenylhydrazone 1a, we performed DFT calculations at the B3LYP/6-311+G(d,p) level using Gaussian 09,9–11 to gain mechanistic insights. We examined 5-membered (path A) and 6-membered ring formation (path B) pathways. The solvent effect of o-xylene was included in the calculation using the IEFPCM method,12 and frequency calculation was performed for each optimized geometry, which yielded a zero-point vibrational energy (ZPE) value. Fig. 1a shows the energy profile obtained for the reaction of hydrazone 1a.13 The sequence of deprotonation and protonation allows facile E/Z isomerization of the N–N bond through the diazene intermediate 1a′. The relative energy of the transition state for the 6-membered ring formation (TS1a-A-E) is not too high (20.2 kcal mol−1) with respect to 1a-A-Z, consistent with the experimental fact that the reaction of hydrazone 1a yielded 2a. Nevertheless, the organopotassium intermediate 2a-A is higher in energy than the reactant state (1a-A-Z) by 19.1 kcal mol−1, which will render the equilibrium between these two states largely in favor of the reactant state. The reason why the yield of 2a became higher in the presence of Et3COH is probably because Et3COH donates a proton to the carbanion 2a-A to facilitate the reaction in the forward direction.14
 | ||
| Fig. 1 (a) Energy diagrams (in kcal mol−1) for the reaction of the deprotonated hydrazone, determined at the B3LYP(IEFPCM)/6-311+G(d,p) level with ZPE corrections. (b) Variation of energy (in kcal mol−1) with a decreasing N–C bond distance, as obtained from relaxed energy calculations. | ||
The DFT calculations further showed that the reaction of hydrazone 1a favors 6-membered ring formation (path B) over the 5-membered ring one (path A). In fact, a local energy minimum corresponding to a 5-membered ring intermediate did not exist on the potential energy surface (see Fig. 1b, path A). This trend is markedly different from that for the oxime substrate, which favors 5-membered ring formation (Fig. S1†).5a In contrast to the case of hydrazone, a 6-membered ring intermediate was not obtained for the oxime substrate (Fig. S3†). To identify the reason why hydrazone 1a selectively undergoes 6-membered ring formation, we inspected the highest occupied molecular orbitals (HOMOs). The HOMO of 1a-A-E is a π-type orbital, which extends perpendicularly to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–N plane, while the HOMO-1 is an in-plane lone-pair type orbital (Fig. 2a). The latter orbital will be mainly responsible for the nucleophilic attack on the alkene carbon. The HOMO-1 has large distributions on both of the two nitrogen atoms of the hydrazone moiety, suggesting that these two nitrogen atoms are almost equally reactive. However, 5-membered ring formation causes a significant steric clash between the phenyl groups on the hydrazone and alkenyl moieties. To prevent this steric clash, the CN–N–Ph moiety undergoes rotation, which disrupts the stabilization gained by conjugation (Fig. 2b, path A). The C–N–N–C torsion angle of this moiety was −138.2° at r = 1.8 Å, indicating a significant loss of planarity. By contrast, the steric clash is much less severe in the 6-membered ring formation, as reflected by the almost unaltered planarity of this moiety at TS1a-A-E (torsion angle = 175.4°, see Fig. 2b, path B). These explain why a 6-membered ring is preferentially formed in the hydroamination of hydrazone 1a.15
N–N plane, while the HOMO-1 is an in-plane lone-pair type orbital (Fig. 2a). The latter orbital will be mainly responsible for the nucleophilic attack on the alkene carbon. The HOMO-1 has large distributions on both of the two nitrogen atoms of the hydrazone moiety, suggesting that these two nitrogen atoms are almost equally reactive. However, 5-membered ring formation causes a significant steric clash between the phenyl groups on the hydrazone and alkenyl moieties. To prevent this steric clash, the CN–N–Ph moiety undergoes rotation, which disrupts the stabilization gained by conjugation (Fig. 2b, path A). The C–N–N–C torsion angle of this moiety was −138.2° at r = 1.8 Å, indicating a significant loss of planarity. By contrast, the steric clash is much less severe in the 6-membered ring formation, as reflected by the almost unaltered planarity of this moiety at TS1a-A-E (torsion angle = 175.4°, see Fig. 2b, path B). These explain why a 6-membered ring is preferentially formed in the hydroamination of hydrazone 1a.15
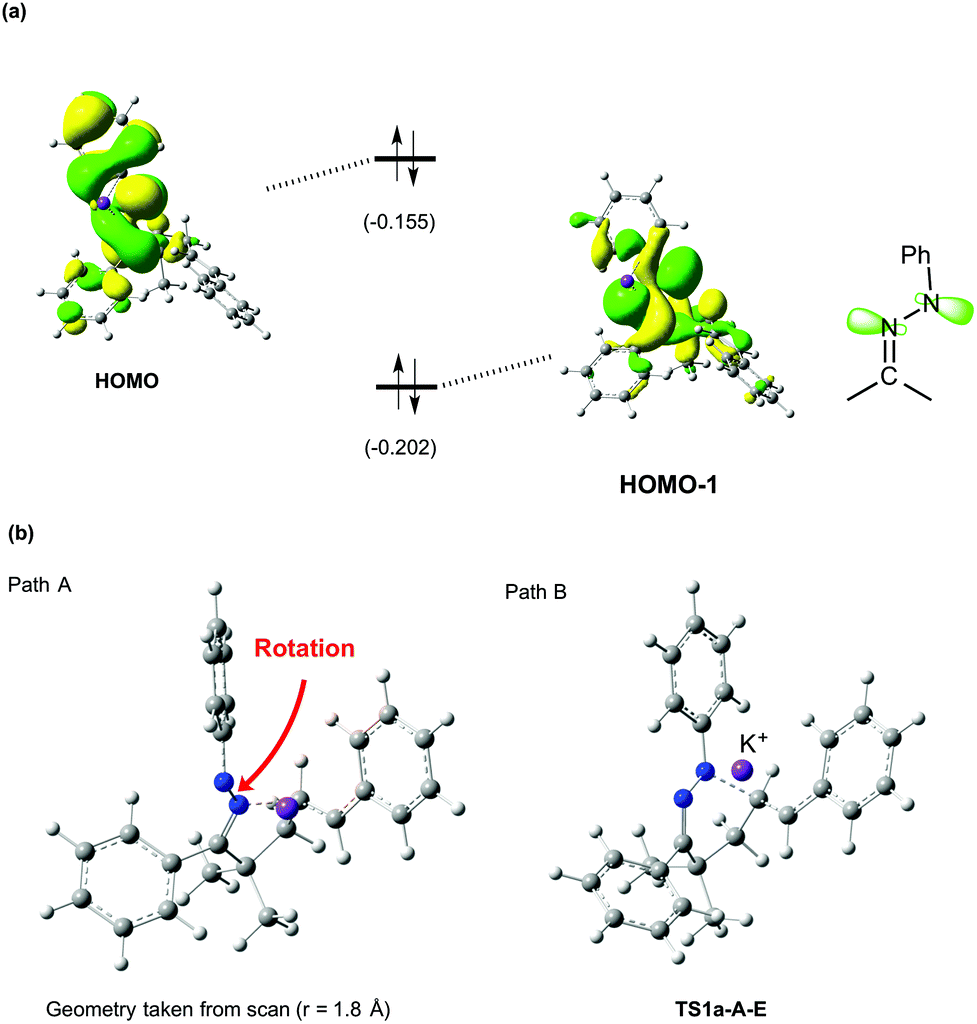 | ||
| Fig. 2 (a) HOMO and HOMO-1 of 1a-A-E. The values in parentheses are the orbital energy levels in hartrees. (b) Geometries of 1a-A-Z at r = 1.8 Å (left) and TS1a-A-E (right). | ||
Having obtained the optimal reaction conditions for hydroamination of hydrazone 1a (Table 1, entry 2) and an understanding of the reaction mechanism, we next examined the substrate scope using a variety of hydrazones 1 (Scheme 2). The reactions with varying R1 allowed for the installation of not only aryl/heteroaryl units (for 2b–d) but also a methyl group (for 2e), while maintaining good product yields. We then turned our attention to the substituents on the alkene (R2 and R3). The steric effects did not influence the reaction outcome for hydroamination of aryl alkenes (for 2f and 2g). Hydroamination with even an electron-rich aryl alkene (for 2h) proceeded smoothly. The reaction of hydrazone 1i having the trisubstituted E-alkene with Ph as R2 and Me as R3 did not retain alkene stereochemistry in the present hydroamination step, affording a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture of 2i. This result implies that the present hydroamination proceeds in a stepwise manner via the carbanion intermediate like 2a-A in Fig. 1. The reaction of N-(para-methoxyphenyl)hydrazone 1j gave a good yield of the hydroamination product 2j. Spirocyclic structures could be constructed in moderate to good yields (for 2k and 2l). The reactions of 1m and 1n having α-proton(s) proceeded well to give the corresponding tetrahydropyridazines 2m and 2n in 76% and 81% yields, respectively.
:1 diastereomeric mixture of 2i. This result implies that the present hydroamination proceeds in a stepwise manner via the carbanion intermediate like 2a-A in Fig. 1. The reaction of N-(para-methoxyphenyl)hydrazone 1j gave a good yield of the hydroamination product 2j. Spirocyclic structures could be constructed in moderate to good yields (for 2k and 2l). The reactions of 1m and 1n having α-proton(s) proceeded well to give the corresponding tetrahydropyridazines 2m and 2n in 76% and 81% yields, respectively.
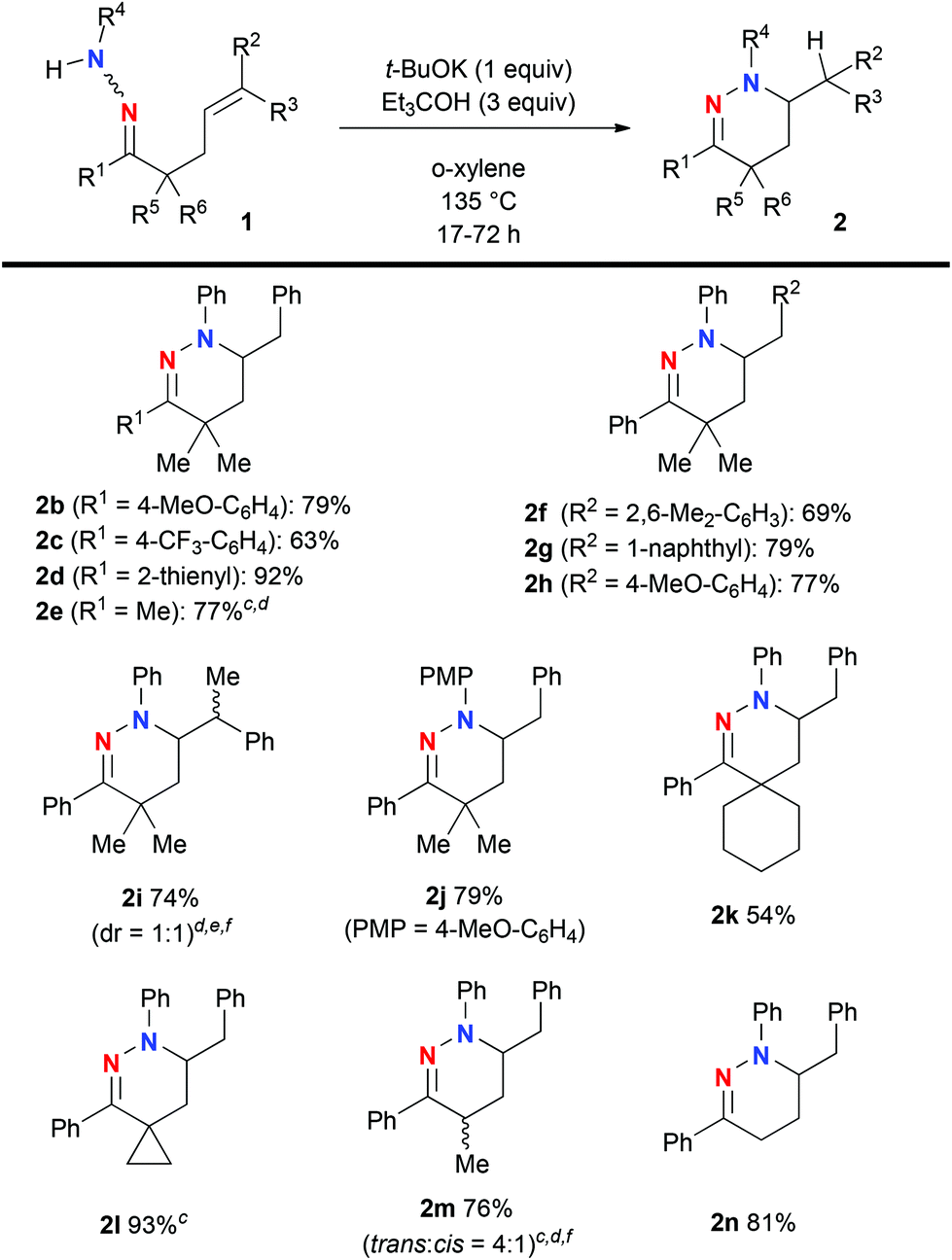 | ||
| Scheme 2 a Unless otherwise noted, the reactions were carried out on the scale of 0.5 mmol of hydrazones 1 in o-xylene (5 mL) under an Ar atmosphere. b Isolated yields were recorded. c The 2-step yield from ketone (0.5 mmol) via hydrazone formation and hydroamination was noted. See the ESI† for more details. d The reaction was conducted in the presence of t-BuOH (10 equiv.) instead of Et3COH. e The reaction was conducted using 0.3 mmol of 2i. f The diastereomeric ratio was judged by 1H NMR analysis of the isolated mixture of 2 and shown in parentheses. | ||
The present hydroamination of hydrazones was applied to the functionalization of the terminal alkene 1o, which gave tetrahydropyridazine 2o and dihydropyrazole 3o in 39% and 8% yields, respectively, along with 21% recovery of 1o (Scheme 3a). Dihydropyrazole 3o was formed presumably via 5-exo cyclization of β,γ-unsaturated hydrazone 1o′, which is generated via alkene isomerization from the terminal to the internal under the present reaction conditions. Indeed, the present hydroamination method enabled us to construct dihydropyrazole 3p in 90% yield via 5-exo hydroaminative cyclization of β,γ-unsaturated hydrazone 1p (Scheme 3b).
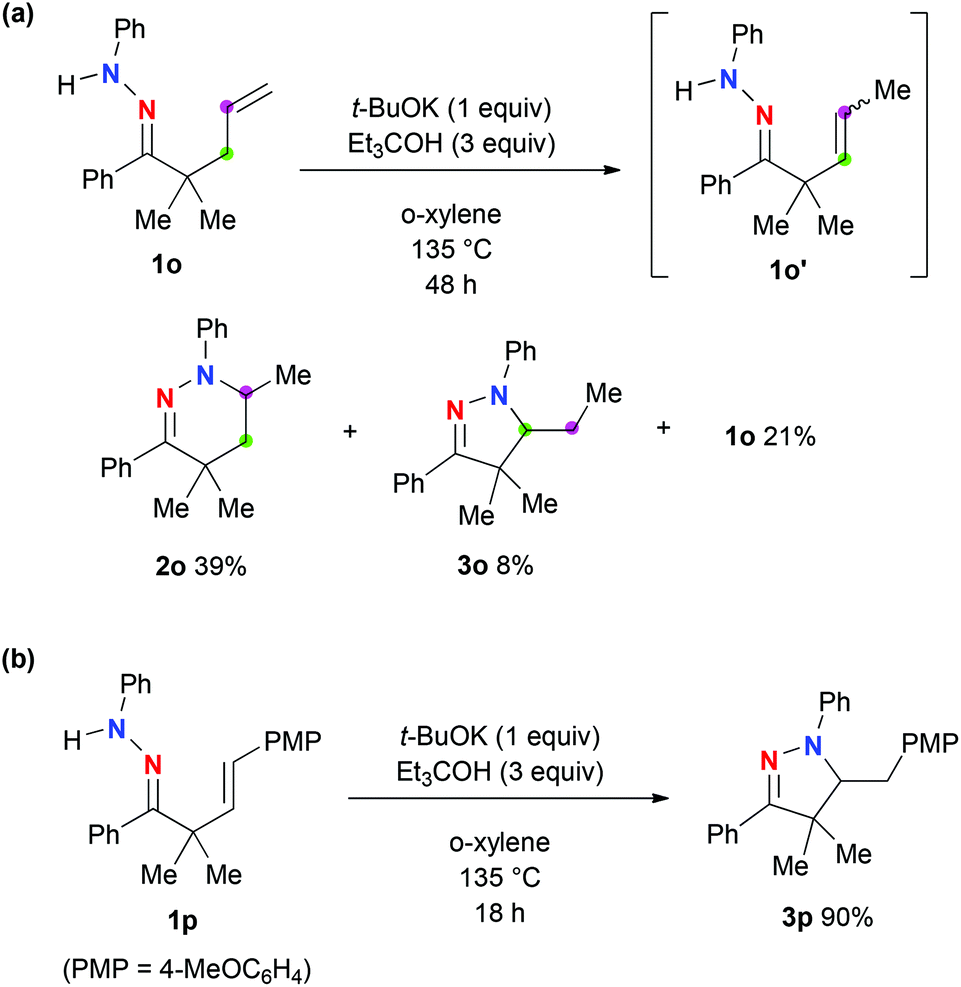 | ||
| Scheme 3 a The reactions were carried out on the scale of 0.5 mmol of hydrazones 1 in o-xylene (5 mL) under an Ar atmosphere. b Isolated yields were recorded above. | ||
In summary, we have developed t-BuOK-mediated hydroamination of alkenyl N-arylhydrazones mainly for the synthesis of tetrahydropyridazine derivatives. Hydrazones have exhibited a wide spectrum of chemical reactivity owing to their unique chemical structure.16 In the area of azaheterocycle synthesis, hydrazones have been typically utilized in the Fischer indole synthesis17 and as a precursor of azomethine imine 1,3-dipoles for [3 + 2]-cycloaddition with dipolarophiles.18,19 The present work offers a new reaction entry of hydrazones towards azaheterocycle synthesis, which is enabled by a simple operation with t-BuOK.
This work was supported by funding from Nanyang Technological University (NTU) and the Singapore Ministry of Education (Academic Research Fund Tier 2: MOE2012-T2-1-014). H. H. thanks the High Performance Computing Centre of NTU for computer resources.
Notes and references
- For general reviews, see: (a) L. D. Quin and J. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, John Wiley & Sons Inc., New York, 2010 Search PubMed; (b) Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Pergamon, Oxford, 2008 Search PubMed; (c) Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, E. F. V. Scriven and C. W. Rees, Pergamon, Oxford, 1996 Search PubMed; (d) Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon, Oxford, 1984 Search PubMed; (e) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles, Wiley-VCH, Weinheim, 2003 CrossRef.
- For selected reviews, see: (a) G. L. Thomas and C. W. Johannes, Curr. Opin. Chem. Biol., 2011, 15, 516 CrossRef CAS PubMed; (b) R. Tohme, N. Darwiche and H. Gali-Muhtasib, Molecules, 2011, 16, 9665 CrossRef CAS PubMed; (c) S. Dandapani and L. A. Marcaurelle, Curr. Opin. Chem. Biol., 2010, 14, 362 CrossRef CAS PubMed; (d) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed; (e) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC.
- For general reviews on hydroamination, see: (a) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed; (b) E. Bernoud, C. Lepori, M. Mellah, E. Schulz and J. Hannedouche, Catal. Sci. Technol., 2015, 5, 2017 RSC; (c) J. Hannedouche and E. Schulz, Chem. – Eur. J., 2013, 19, 4972 CrossRef CAS PubMed; (d) P. S. Hanley and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 8510 CrossRef CAS PubMed; (e) K. D. Hesp and M. Stradiotto, ChemCatChem, 2010, 2, 1192 CrossRef CAS; (f) T. E. Mülller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed; (g) R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407 RSC; (h) S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed; (i) J. Seayad, A. Tillack, C. G. Hartung and M. Beller, Adv. Synth. Catal, 2002, 344, 795 CrossRef CAS; (j) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef.
- Retro-Cope type hydroamination (1,3-azaprotio cyclotransfer) with hydroxyl amine and hydrazine derivatives is a powerful but mechanistically distinct alternative that proceeds via a concerted pericyclic mechanism whereby C–N and C–H bonds are formed concurrently. For reviews, see: (a) A. M. Beauchemin, Org. Biomol. Chem., 2013, 11, 7039 RSC; (b) N. J. Cooper and D. W. Knight, Tetrahedron, 2004, 60, 243 CrossRef CAS.
- (a) X. Peng, B. M. K. Tong, H. Hirao and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 1959 CrossRef CAS PubMed; (b) A. Kaga, X. Peng, H. Hirao and S. Chiba, Chem. – Eur. J., 2015, 21, 19112 CrossRef CAS PubMed.
- Retro-Cope intramolecular hydroamination of alkynyl hydrazides delivers cyclic azomethineimines, see: A. D. Hunt, I. Dion, N. Das Neves, S. Taing and A. M. Beauchemin, J. Org. Chem., 2013, 78, 8847 CrossRef CAS PubMed.
- For reports on retro-Cope intramolecular hydroamination of alkenyl hydrazides, see: (a) C. Clavette, J.-F. V. Rocan and A. M. Beauchemin, Angew. Chem., Int. Ed., 2013, 52, 12705 CrossRef CAS PubMed; (b) F. Loiseau, C. Clavette, M. Raymond, J.-G. Roveda, A. Burrell and A. M. Beauchemin, Chem. Commun., 2011, 47, 562 RSC; (c) J.-G. Roveda, C. Clavette, A. D. Hunt, S. I. Gorelsky, C. J. Whipp and A. M. Beauchemin, J. Am. Chem. Soc., 2009, 131, 8740 CrossRef CAS PubMed.
- For a report on t-BuOK-catalzyed hydroamination of aryl alkenes, see: M. Beller, C. Breindl, T. H. Riermeier, M. Eichberger and H. Trauthwein, Angew. Chem., Int. Ed. Engl., 1998, 37, 3389 CrossRef CAS . Also see ref. 3i.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS; (c) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- The stereochemistry of the hydrazone N–N bond does not influence the outcome of the present hydroamination, as E/Z-isomerization of the N–N bond occurs readily under the present reaction conditions.
- Prior deprotonation of hydrazone 1a is essential for the cyclization reaction, because the reaction of neutral hydrazone 1a has a very high energy barrier (see the ESI, Fig. S2†).
- On the other hand, the HOMO of the deprotonated oxime is an in-plane orbital (see the ESI, Fig. S4†). The HOMO lobe is highly localized on the nitrogen, especially on the side of new bond formation, which is beneficial for 5-membered ring formation. However, the HOMO lobe on the oxygen atom looks like a pure p-orbital and is nearly perpendicular to the N–O bond. Thus, the oxygen may not be able to interact with the alkene effectively (Fig. S5b†). These characteristics of the HOMO seem to provide a rationale for the observation that the formation of a 5-membered ring is preferred in the oxime case.
- For general reviews, see: (a) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS PubMed; (b) A.-Z. A. Elassar, H. H. Dib, N. A. Al-Awadi and M. H. Elnagdi, ARKIVOC, 2007, ii, 272 Search PubMed; (c) H. R. Vollmer, Synlett, 1999, 1844 CrossRef CAS; (d) R. O. Hutchins and M. K. Hutchins, in Comprehensive Organic Synthesis, ed. B. M. Trost, Elsevier, Oxford, 1991, vol. 8, p. 327 Search PubMed.
- For reviews, see: (a) M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29 RSC; (b) B. Robinson, Chem. Rev., 1969, 69, 785 CrossRef CAS PubMed; (c) B. Robinson, Chem. Rev., 1969, 69, 227 CrossRef CAS; (d) R. R. Phillips, Org. React., 1959, 10, 1143 Search PubMed.
- (a) R. Grigg, Chem. Soc. Rev., 1987, 16, 89 RSC; (b) X. Deng and N. S. Mani, J. Org. Chem., 2008, 73, 2412 CrossRef CAS PubMed.
- For the synthesis of cyclic azomethine imines possessing a β-aminocarbonyl motif via alkene aminocarbonylation of hydrazones, see: (a) K. Lavergne, A. Bongers, L. Betit and A. M. Beauchemin, Org. Lett., 2015, 17, 3612 CrossRef CAS PubMed; (b) W. Gan, P. J. Moon, C. Clavette, N. Das Neves, T. Markiewicz, A. B. Toderian and A. M. Beauchemin, Org. Lett., 2013, 15, 1890 CrossRef CAS PubMed; (c) C. Clavette, W. Gan, A. Bongers, T. Markiewicz, A. Toderian, S. I. Gorelsky and A. M. Beauchemin, J. Am. Chem. Soc., 2012, 134, 16111 CrossRef CAS PubMed; (d) D. W. Jones, J. Chem. Soc., Chem. Commun., 1982, 766 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, including procedures, syntheses and characterization of new compounds; 1H and 13C NMR spectra. CCDC 1001827–1001792. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00053c |
| This journal is © the Partner Organisations 2016 |