
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in engineering nonribosomal peptide assembly lines
M.
Winn
,
J. K.
Fyans
,
Y.
Zhuo
and
J.
Micklefield
*
School of Chemistry and Manchester Institute of Biotechnology, The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. E-mail: Jason.micklefield@manchester.ac.uk
First published on 24th December 2015
Abstract
Covering: up to July 2015
Nonribosomal peptides are amongst the most widespread and structurally diverse secondary metabolites in nature with many possessing bioactivity that can be exploited for therapeutic applications. Due to the major challenges associated with total- and semi-synthesis, bioengineering approaches have been developed to increase yields and generate modified peptides with improved physicochemical properties or altered bioactivity. Here we review the major advances that have been made over the last decade in engineering the biosynthesis of nonribosomal peptides. Structural diversity has been introduced by the modification of enzymes required for the supply of precursors or by heterologous expression of tailoring enzymes. The modularity of nonribosomal peptide synthetase (NRPS) assembly lines further supports module or domain swapping methodologies to achieve changes in the amino acid sequence of nonribosomal peptides. We also review the new synthetic biology technologies promising to speed up the process, enabling the creation and optimisation of many more assembly lines for heterologous expression, offering new opportunities for engineering the biosynthesis of novel nonribosomal peptides.
 M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield | Michael Winn earned his PhD in 2012 with Rebecca Goss at the University of East Anglia, before spending a year at University College Dublin with Cormac Murphy. He has been working with Jason Micklefield since 2013. Joanna Fyans obtained her PhD in 2011 on molecular microbiology and plant pathology with Prof. Tracy Palmer at the University of Dundee and Prof. Ian Toth at the James Hutton Institute, before moving to focus on phytotoxins produced by Streptomyces with Prof. Dawn Bignell at Memorial University. Ying Zhuo received her PhD in biology and biochemistry from Institute of Microbiology, Chinese Academy of Sciences, in 2010. They both joined the Micklefield group in 2014. Jason Micklefield graduated from the University of Cambridge in 1993 with a PhD in Chemistry, with Prof. Sir Alan R. Battersby FRS. He then moved to the University of Washington as a NATO postdoctoral fellow working with Prof. Heinz G. Floss. In 1995 he became a lecturer in organic chemistry at Birkbeck College before moving to the University of Manchester in 1998 where he is Professor of Chemical Biology within the School of Chemistry and the Manchester Institute of Biotechnology. His research interests include biosynthesis, biocatalysis and riboswitches. |
1 Introduction
Nonribosomal peptides are amongst the most widespread and structurally diverse secondary metabolites in nature, possessing a broad range of biological activities which have been exploited in the development of a variety of important therapeutic agents such as the immunosuppressant cyclosporine A, the antibiotic daptomycin, or the anticancer bleomycin A2 (Fig. 1). The structural complexity of many nonribosomal peptides renders total synthesis impractical and semi-synthesis challenging, although there have been several examples of semi-synthesis being performed successfully, such as the vancomycin-based Oritavancin which was approved by the FDA in 2014 for treatment of drug resistant skin infections.1 Consequently there is major interest in the development of bioengineering approaches that increase the yields of nonribosomal peptides and in the generation of modified peptides with altered bioactivity or improved physicochemical properties.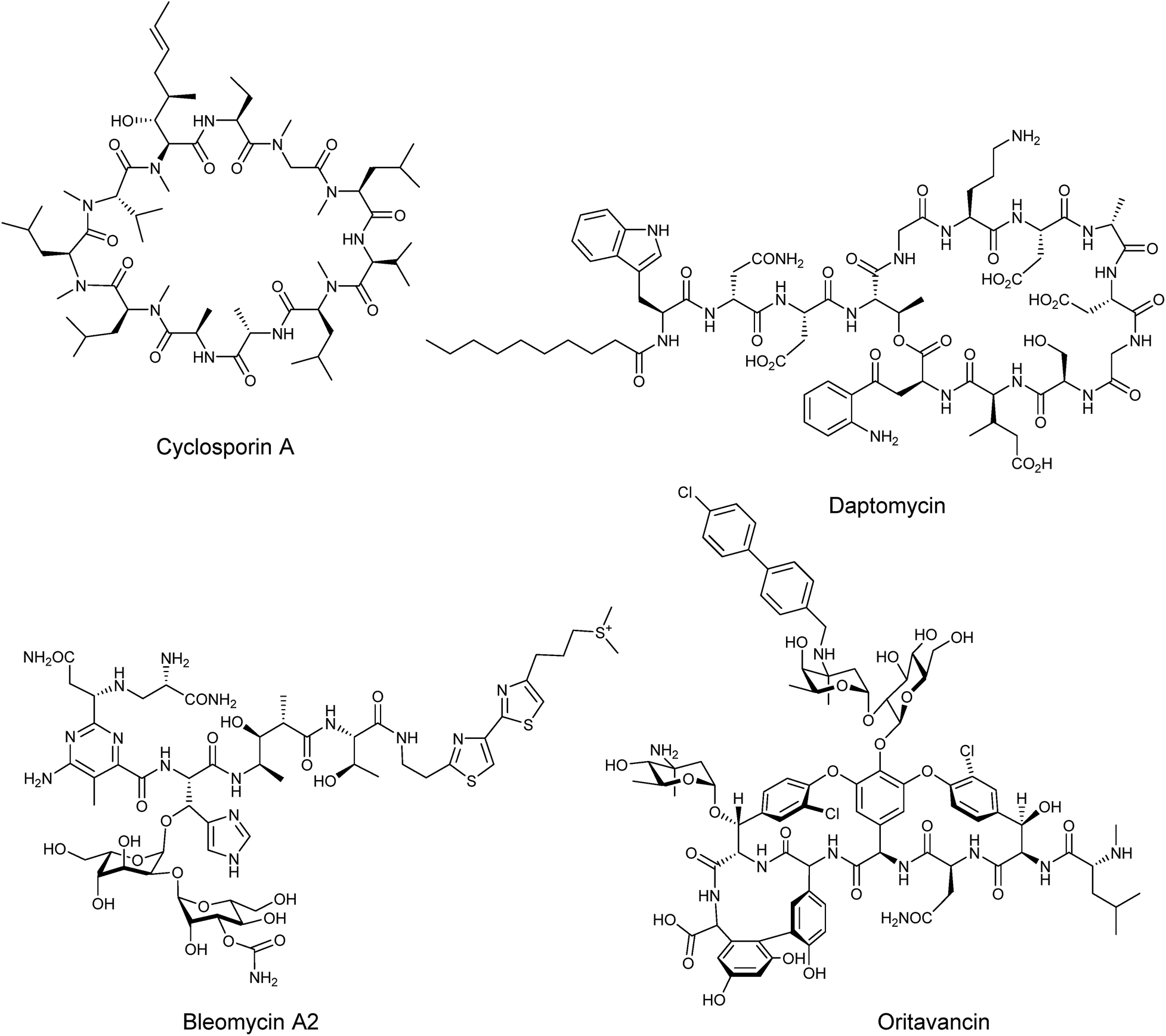 | ||
| Fig. 1 Structures of some clinically relevant nonribosomal peptides. | ||
Nonribosomal peptides are biosynthesised by large, modular, multifunctional enzymes known as nonribosomal peptide synthetases (NRPS) (Fig. 2). Each module within an NRPS is responsible for the incorporation of a single building block into the final polypeptide structure. Since every incorporated amino acid requires a specific module, nonribosomal peptide synthetases can be extremely large enzymes. For example, the single NRPS responsible for cyclosporine A assembly in Tolypocladium niveum is 1.6 MDa in size.2 In general, NRPS modules in bacteria tend to be distributed over a number of smaller subunit proteins which associate into a larger multi-enzyme system.
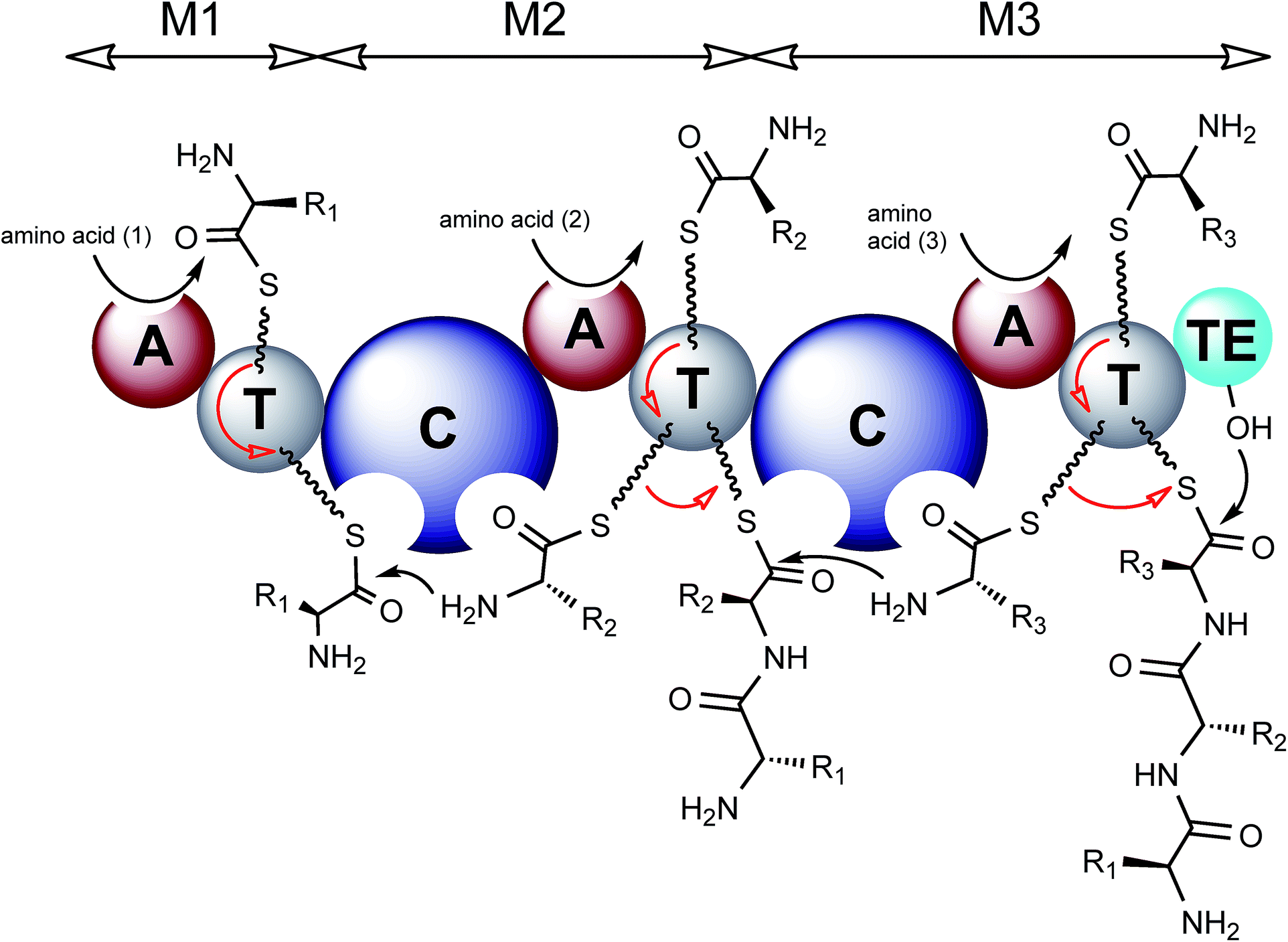 | ||
| Fig. 2 Model of NRPS biosynthesis. Amino acid substrates are activated through reaction with ATP to form an aminoacyl-AMP intermediate, catalysed by an adenylation domain (A). The aminoacyl-AMP intermediate is then captured by the thiol group of the flexible 4′-phosphopantetheine arm tethered to a thiolation domain (T). Condensation domains (C) catalyse successive peptide bond formation between the thioester intermediates loaded onto adjacent T domains. The first module is known as the initiation module (M1) and subsequent modules are known as elongation modules. Each module incorporates a single amino acid, therefore there are as many modules required as there are amino acids in the final peptide product. The final module contains an additional thioesterase domain (TE) which catalyses hydrolysis or cyclisation to release the peptide from the NRPS. Modules may contain additional domains including epimerisation (E), N-methylation (NMT) and cyclisation domains (Cy). The released peptide can subsequently be modified by tailoring enzymes, further increasing structural diversity. | ||
Major insights into the substrate specificity of NRPS domains came when the first structure of an adenylation (A) domain was determined. The structure of the phenylalanine activating A domain from GrsA, an NRPS involved in gramicidin S synthesis, was solved in complex with AMP and L-phenylalanine.3 In this structure the active site residues, responsible for binding the substrate Phe, were identified thus enabling the NRPS specificity code to be deciphered. This allows the prediction, with fairly high levels of accuracy, of the cognate substrate of a module.4,5 In addition to the 21 proteinogenic amino acids, NRPS modules can also incorporate unusual, non-proteinogenic, amino acids including D-amino acids. Hybrid NRPS assembly lines are also known which include polyketide synthase (PKS) and other enzyme activities.6
The first module in an NRPS is known as the initiation module and can typically be subdivided into an adenylation domain (A) and a thiolation domain (T), also known as a peptidyl carrier protein domain (PCP). Following this are a number of elongation modules which also contain A and T domains but have an additional upstream condensation domain (C). The cycle of nonribosomal peptide synthesis requires the priming of a conserved serine residue within the T domain by the addition of a flexible 4′-phosphopantetheine (PPT) prosthetic group, catalysed by a 4′-phosphopantetheinyl transferase (PPTase). This flexible linker allows tethered intermediates to be passed from one domain to another along the assembly line. Following the priming of the PCP, the A domain of the initiation module activates its cognate amino acid substrate through a reaction with ATP to generate an aminoacyl-AMP intermediate which is attacked by the thiol group of the PPT resulting in a PCP-tethered aminoacyl thioester (Fig. 2). The A domain of module 2 similarly activates its amino acid substrate to generate a second aminoacyl thioester tethered to the PCP of module 2. The condensation domain of module 2 then catalyses peptide formation to give a dipeptide intermediate tethered to the second PCP domain (Fig. 2). The initiation module can then load another substrate amino acid and commence assembly of another peptide. The peptidyl-thioester intermediate is passed from one module to the next with a single amino acid being added at each module. Finally the full length polypeptide is released by a terminating thioesterase (TE) domain which either hydrolyses the linear product or catalyses cyclisation during the release (Fig. 2).7 In addition to these standard modules, further structural variation can be introduced by other optional domains such as epimerization (E), methylation (MT) and cyclization domains (Cy). Epimerization domains occur at the C-terminal end of modules responsible for D-amino installation and act on the PCP-tethered peptide.7 As the product of these domains is a racemic mixture, the C-domain of the downstream module ensures that the correct enantiomer/diastereomer is subsequently used for elongation.8,9 Methylation of nonribosomal peptides is achieved by specialised methylation domains or by standalone enzymes that come in three different flavours (N-, C- or O-methyltransferases) that utilise S-adenosylmethionine as the methyl donor. N-Methyltransferases are most commonly found as domains inserted within the adenylation domain and typically methylate the PCP-tethered amino acid substrate prior to condensation, such as in thaxtomin A biosynthesis.10 Alternatively methylation is catalysed by separate enzymes within the cluster which act in trans on the final, often cyclised, peptide such as chloroeremomycin.11C-Methyltransferases are much more commonly found in PKS rather than NRPS clusters but an example can be found in the yersiniabactin biosynthetic cluster, a hybrid NRPS/PKS from Yersinia pestis, here the methylation domain, found within a nonribosomal peptide module, catalyses methylation of a thiozolinyl-S-PCP intermediate. O-Methylation events are rarer still but an example can be seen within the NRPS cluster for saframycin Mx1 biosynthesis.12 Cyclisation domains (Cy) are unusual tailoring enzymes as they take the place of the condensation domain in a module and catalyse the formation of a peptide bond via the heterocyclisation of cysteine, serine and threonine residues to thiazoline or oxazoline heterocycles. In many cases the resulting heterocycle is then oxidised by an oxidase domain (Ox) to the corresponding thiazole, for example during epothilone biosynthesis.13 More detailed explanations of tailoring domains and their functions have been covered in previous reviews.7,14
Although nonribosomal peptides can have an important function in the producing organism, such as iron-scavenging carried out by siderophores,15 most interest in these molecules relates to the fact that nonribosomal peptides display a wide range of bioactivities; nonribosomal peptides can be exploited in agrochemical applications, or in the development of therapeutic agents including anti-tumour, antiviral, immunosuppressive and antimicrobial agents. Although many nonribosomal peptides exhibit significant biological activity many do not possess desirable pharmacokinetics or ADME properties and so the semi-synthesis or engineered biosynthesis of nonribosomal peptide variants is desirable. With the emergence of antibiotic resistance among pathogenic bacteria, there is currently massive interest in developing new and more effective antimicrobial agents. Early researchers in this field envisioned that the assembly lines of nonribosomal peptides could be engineered to incorporate different residues thereby producing new and improved “non-natural” products. This review seeks to cover the progress in engineering nonribosomal peptides that has occurred in the last ten years.
2 Early developments: precursor directed biosynthesis and mutasynthesis
Much of the early work into the biosynthetic generation of novel natural product analogues focused on precursor directed biosynthesis (PDB) or mutasynthesis, two terms for methods that differ slightly and are oftentimes, inaccurately, used interchangeably. In precursor directed biosynthesis a wild-type nonribosomal peptide-producing organism is provided with modified or synthetic amino acids with the expectation that the substrate specificity of the relevant NRPS is flexible enough to allow incorporation of the modified precursors into the final peptide. The incorporation of the modified amino acid often occurs in competition with the native building blocks, leading to production of a mixture of wild-type and modified product. One advantage of the precursor directed approach is that it requires only a limited understanding of the biosynthetic machinery and as a result there are many early examples of its use to produce nonribosomal peptide analogues. For example, novel cyclosporin analogues were produced in the 1980s by feeding various un-natural amino acids to cultures of the cyclosporin producer Tolypocladium inflatum. As a result, cyclosporin variants (1), (2) and (3) were produced through the incorporation of unnatural precursors allylglycine, β-cyclohexylalanine or D-serine respectively. Notably, the cyclosporin analogue (3), with D-serine in place of the natural D-alanine at position 8, exhibited high levels of biological activity (Fig. 3).16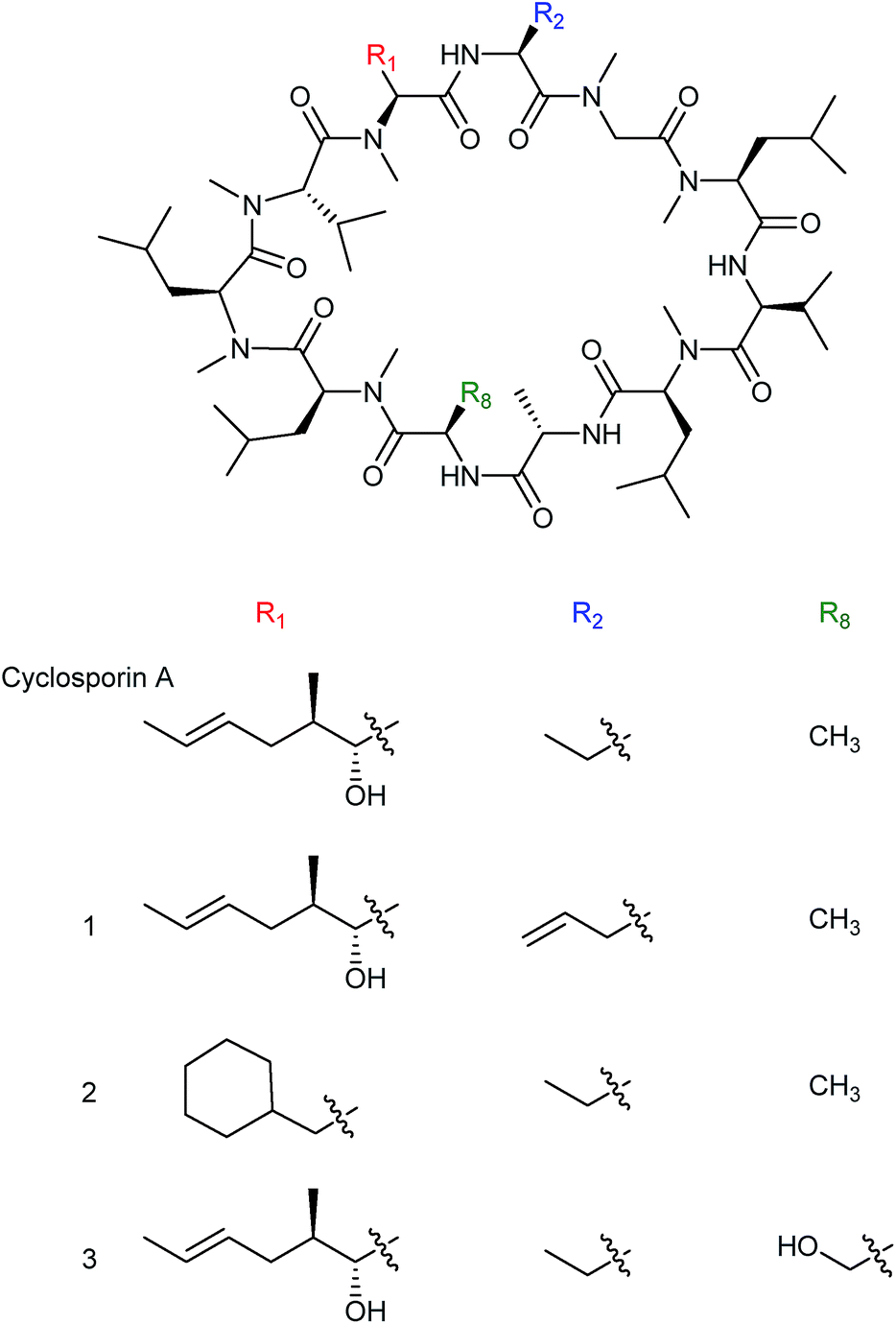 | ||
| Fig. 3 Cyclosporin analogues incorporating nonnatural allylglycine (1), β-cyclohexylalanine (2) or D-serine residues (3) produced by precursor directed biosynthesis.16 | ||
Many other examples of precursor directed biosynthesis can be found in earlier reviews.17 Although there are many examples where precursor directed biosynthesis has been used effectively, one of the problems associated with this technique is that the synthetic precursors compete with the natural endogenous amino acid precursors, which likely act as the preferred substrates. As a result the isolated yields of novel compounds can be low with the wild-type products predominant. As a solution to this problem, mutasynthesis was developed. In this process the modified substrates are fed to an engineered organism which is deficient in the enzyme(s) required for the biosynthesis of a specific natural precursor, so that a precursor analogue may be more effectively incorporated. In contrast to precursor directed biosynthesis, a reasonable amount of genetic information has to be known about the biosynthetic gene cluster and the genetic tractability of the producing organism.
Using a mutasynthesis approach, novel calcium-dependent antibiotics (CDAs) were generated through the creation of a Streptomyces coelicolor strain where the production of CDA was abolished following deletion of the gene hmaS. This gene is involved in the biosynthesis of 4-hydroxymandelic acid, a precursor for the biosynthesis of 4-hydroxyphenylglycine (L-Hpg), which is one of the non-proteinogenic amino acids installed in the CDA structure (Fig. 4).18 A series of novel lipopeptides were produced when the mutant was instead supplied with a number of synthetic mandelate, arylglyoxylate and arylglycine analogues. Feeding of phenylglycine instead of L-Hpg led to CDA variant (4) being produced that lacked the hydroxy group relative to the wild-type CDA. More interestingly, cultures fed with 4-fluorophenylglycine or similarly 4-fluoromandelic acid or 4-fluorophenylglyoxylate, led to the detection of fluorinated analogues (5) and (6). However, similar L-Hpg analogues carrying the bulkier chlorine or methoxy functionalities did not lead to new products.
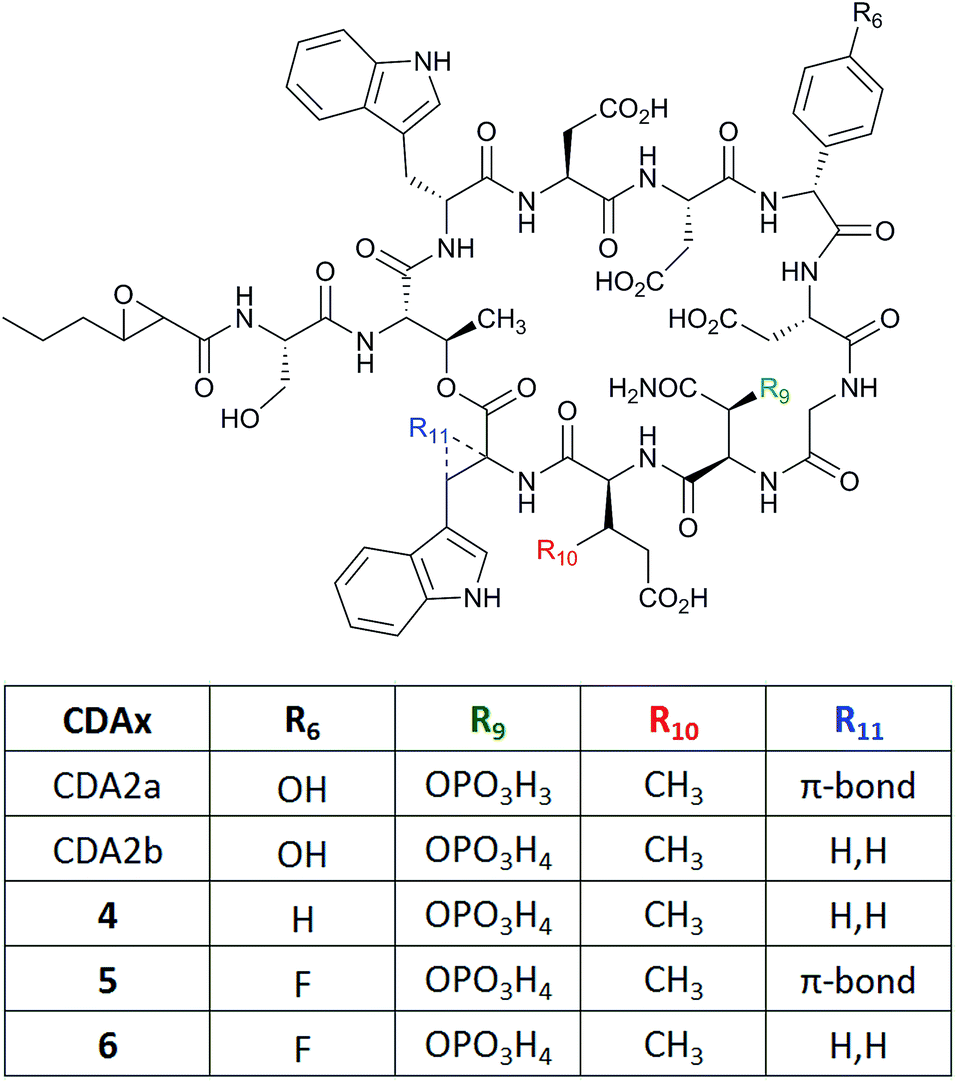 | ||
| Fig. 4 Structures of calcium-dependent antibiotics CDA2a and 2b and variants (4–6) produced by mutasynthesis.18 | ||
In a similar study, the biosynthetic pathway of the vancomycin-related glycopeptide balhimycin was manipulated so that the gene responsible for the formation of the naturally incorporated β-hydroxytyrosine, bhp, was inactivated. Cultures of this deletion mutant were fed with either 2-fluoro-β-hydroxytyrosine (7), 3-fluoro-β-hydroxytyrosine (8) or 3,5-difluoro-β-hydroxytyrosine (9) to yield the corresponding fluorinated balhimycins (10), (11) and (12) (Fig. 5).19 As with the previous example, not all the tested β-hydroxytyrosine analogues led to novel glycopeptide structures; several β-hydroxytyrosine analogues lacking the para-hydroxyl group failed to be incorporated.
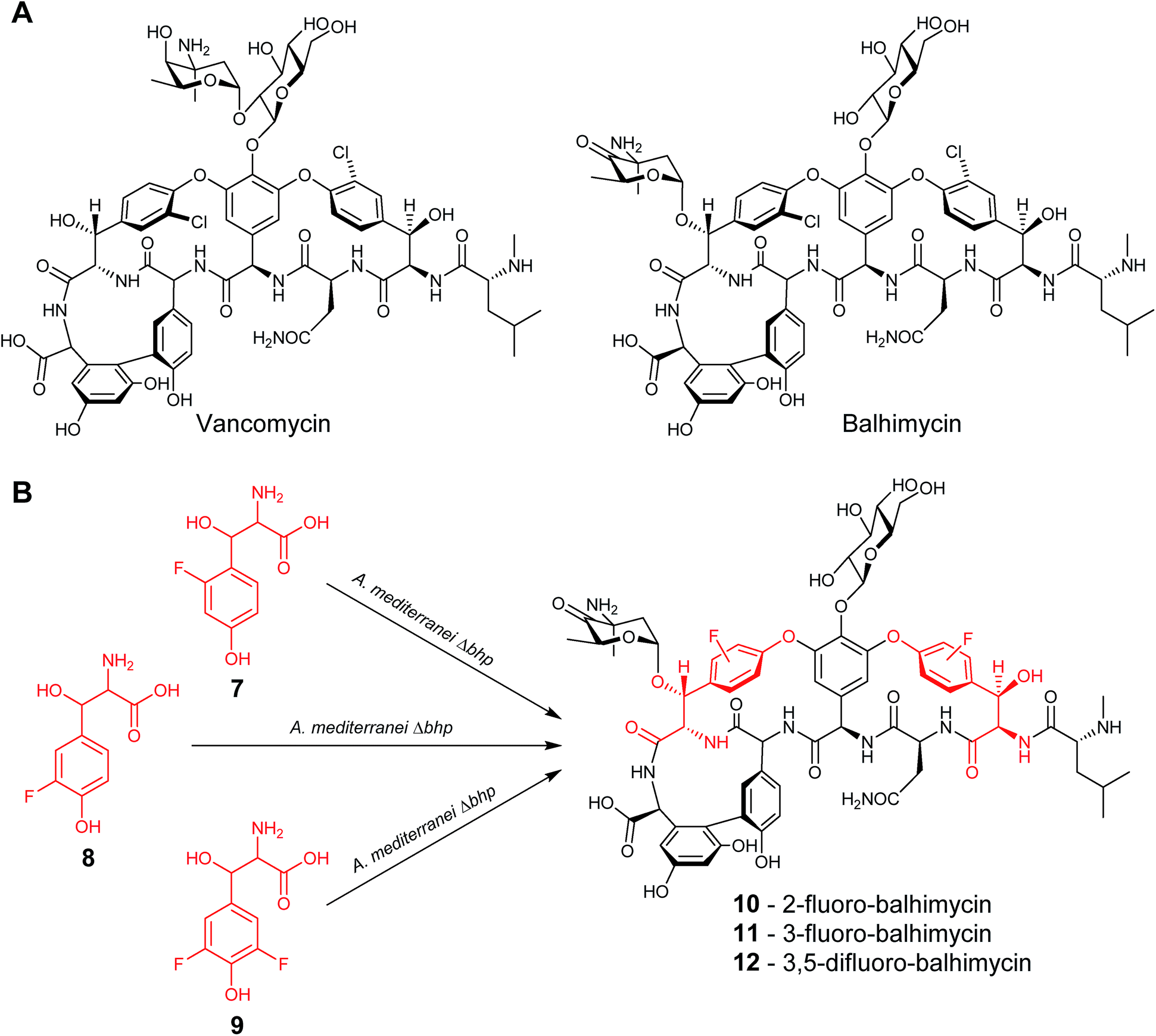 | ||
| Fig. 5 (A) Structures of the glycopeptides vancomycin and balhimycin. (B) Feeding of fluorinated β-hydroxytyrosine led to the isolation of the correspondingly fluorinated balhimycins.19 | ||
These two examples help to highlight a recurring problem in the traditional mutasynthesis and precursor directed biosynthesis approaches in that the introduced changes are usually conservative due to limited or uncompromising substrate flexibility of the native enzymes. Moreover, these examples were limited to modifications of non-proteinogenic amino acids as gene deletions that abolish production of these non-essential amino acids do not, on the whole, affect growth. Introducing modifications to the proteinogenic amino acid residues can be more challenging, requiring the creation of amino acid auxotrophs and feeding experiments conducted in minimal media.20
Although the techniques of precursor directed biosynthesis and mutasynthesis have been utilized for some time they are still in regular use as they offer a simple means of generating functional analogues of nonribosomal peptides.21–24 However recent developments in synthetic biology are opening up new avenues for introducing structural diversity into nonribosomal peptides.
3 Engineering of precursor supply and tailoring enzymes
3.1 Engineering precursor supply
In recent years the principles of synthetic biology are being adopted for the production of new nonribosomal peptides, altering the precursor supply in vivo or introducing tailoring enzymes from other pathways to create structural diversity. Altering precursor supply operates in a similar way to precursor directed biosynthesis and relies on generating altered amino acids prior to their incorporation into the final structure, but with the focus being on endogenous biosynthesis rather than exogenous feeding.The introduction of halogen substituents into nonribosomal peptide scaffolds has been a common target, as simple changes in halogenation patterns can have a significant impact on the activity of a compound. For example, when the enzyme PrnA, a flavin-dependent tryptophan-7-halogenase from Pseudomonas fluorescens Pf-5, was expressed alongside the NRPS genes for the uridyl peptide antibiotic pacidamycin, which is produced by Streptomyces coeruleorubidus,25 a new halogenated analogue was generated. The halogenase gene was cloned into the plasmid pIJ10257, which integrates into the streptomyces φBT1 site, and placed under the control of the ermE* constitutive promoter. The new pacidamycin analogue was halogenated at the C-terminal tryptophan moiety (13), with the tryptophan becoming halogenated by PrnA prior to incorporation by the NRPS. This modified analogue was produced as the minor product alongside the wild-type pacidamycin in a typical ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 but the authors note that in some cases the chlorinated product was produced as the dominant species. Chloropacidamycin was isolated at approximately 1 mg per litre, a yield that was comparable to that achieved in their previous precursor directed biosynthesis work with 7-chlorotryptophan.22 This halogenation approach also provided access to a range of new arylated analogues (14–17) via a semi-synthetic Suzuki–Miyaura coupling reaction performed on the purified pacidamycin analogues (Fig. 6).25
:5 but the authors note that in some cases the chlorinated product was produced as the dominant species. Chloropacidamycin was isolated at approximately 1 mg per litre, a yield that was comparable to that achieved in their previous precursor directed biosynthesis work with 7-chlorotryptophan.22 This halogenation approach also provided access to a range of new arylated analogues (14–17) via a semi-synthetic Suzuki–Miyaura coupling reaction performed on the purified pacidamycin analogues (Fig. 6).25
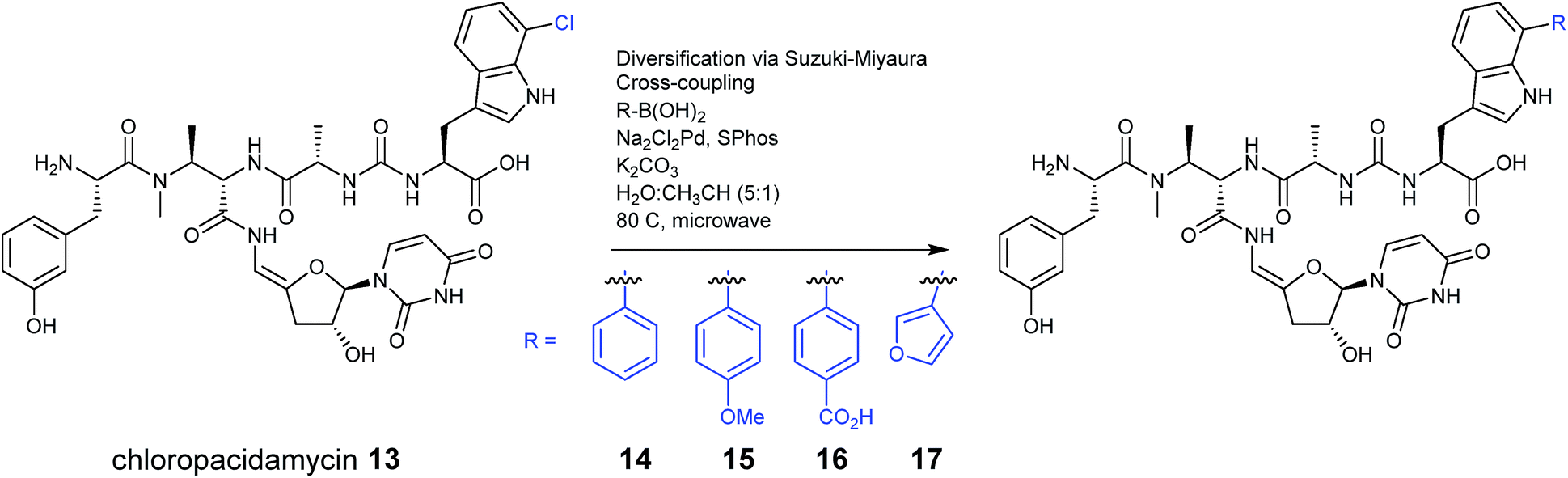 | ||
| Fig. 6 Pacidamycin derivatives were generated by producing 7-chlorotryptophan in vivo, which is subsequently installed at the C-terminus of pacidamycin. Further analogues were then produced using a semi-synthetic approach, using purified pacidamycins.25 | ||
A significant portion of the work on engineering of nonribosomal peptides has focused on the family of lipopeptide antibiotics, an important class of antibiotics that includes the calcium dependent antibiotics, friulimicins and daptomycin. This family of lipopeptides all possess an N-terminal fatty acid chain which aids their penetration into the membrane of Gram-positive bacteria. The length of the fatty acid chain varies between family members and can have a significant impact on antimicrobial activity. The antibacterial activity generally rises with increasing acyl chain length, however chain lengths longer than 11 carbons tend to exhibit toxicity in humans.26,27 Deacylation mechanisms also play a part in increasing resistance to lipopeptide antibiotics, such as daptomycin, so being able to vary this chain offers a route to new effective antibiotic treatments.28 Lewis et al. were able to modify the active site of the β-ketoacyl-ACP synthase FabF3 from Streptomyces coelicolor leading to the installation of fatty acid chains of differing lengths onto CDA.29 CDA has a trans-2,3-epoxyhexanoyl fatty acid side chain, which is unusually short in comparison to most other lipopeptides. The authors first experimented with the fatty acid chain of CDA using more traditional mutasynthesis techniques and determined that the biosynthesis of the CDA lipid moiety is controlled by a fab operon of five genes (Fig. 7B).30 The operon includes a gene encoding an acyl carrier protein (ACP) which facilitates the biosynthesis and transfer of the fatty acid during the first stage of CDA assembly. Also present are genes fabF3 and fabH4 encoding β-ketoacyl-S-ACP synthase enzymes (KAS-II and KAS-III) which catalyse Claisen-type condensation reactions during chain elongation, leading to a hexanoyl-S-ACP intermediate (24). Additionally, there are genes encoding a hexanoyl-ACP oxidase (HxcO), which generates a trans-hexanoyl-S-ACP intermediate (25), and a monooxygenase (HcmO), which catalyses an epoxidation reaction to give the epoxyhexanoyl-ACP (26). Deactivation of the module 1 PCP of CDA biosynthesis prevented the transfer of the upstream ACP-tethered 2,3-epoxyhexanoyl fatty acid chain, therefore production of CDA was abolished. Feeding an exogenous supply of synthetic N-acyl-L-serinyl-NAC analogues restored the production line and allowed detection of CDA analogues with pentanoyl (20) and hexanoyl (21) side chains (Fig. 7A).30
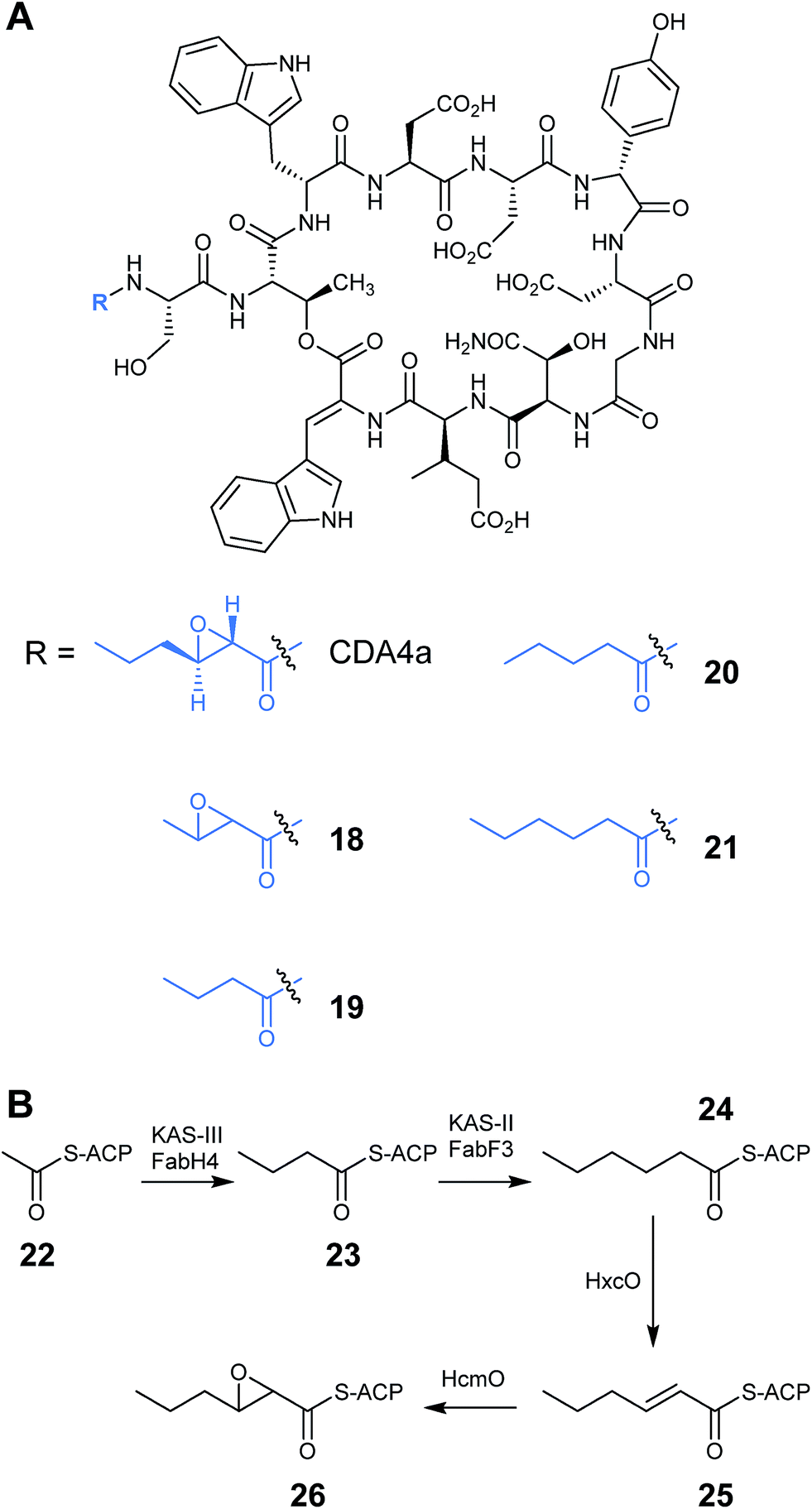 | ||
| Fig. 7 (A) Structure of CDA4a analogues with altered fatty acid side chains. (B) Proposed biosynthesis of CDA epoxyhexanoyl-S-ACP side chain.30 | ||
Sequence analysis of the KAS-II type enzyme, FabF3, showed that the acyl-binding pocket contained a Phe residue at position 107 rather than the smaller amino acids, such as Ile or Leu, which are found in other similar enzymes. The authors speculated that this phenylalanine residue acts as a block to longer chain fatty acids, which explains why CDA contains an unusually short lipid chain. However when mutants were constructed where the Phe107 was replaced with Ile, Leu or Ser, wild-type CDA with the native trans-2,3-epoxyhexanoyl side chain (CDA4a) was still produced rather than CDA products with longer lipid chains. The F107I and F107L mutants did however also produce a small amount of two new products that were identified as being CDA modified with either a 2,3-epoxybutanoyl (18) or a butanoyl (19) fatty acid side chain (Fig. 7A). FabF3 is the second enzyme in the fatty acid chain elongation, which catalyses the condensation of a malonyl unit with butanoyl-S-ACP (23). The fact that CDA analogues were isolated with a butanoyl chain suggested that the FabF3 mutants were lacking in activity compared to the wild-type, leading to the accumulation of the butanoyl intermediate. Nevertheless, the fact that these intermediates were successful in initiating the CDA core peptide, together with the earlier mutasynthesis results, suggested a certain flexibility in the initiation module of CDA. The formation of CDA variants with epoxybutanoyl fatty acids also demonstrated that the epoxide forming monooxygenase also has a certain degree of substrate promiscuity.29 These results could potentially lead to further novel structures being produced with more variation in the fatty acid chain of lipopeptide antibiotics.
3.2 Tailoring enzymes
In addition to altering the nature of the incorporated precursors the structural diversity of nonribosomal peptides can be increased by utilising exogenous tailoring enzymes from other pathways alongside the NRPS machinery to diversify the final peptide structure.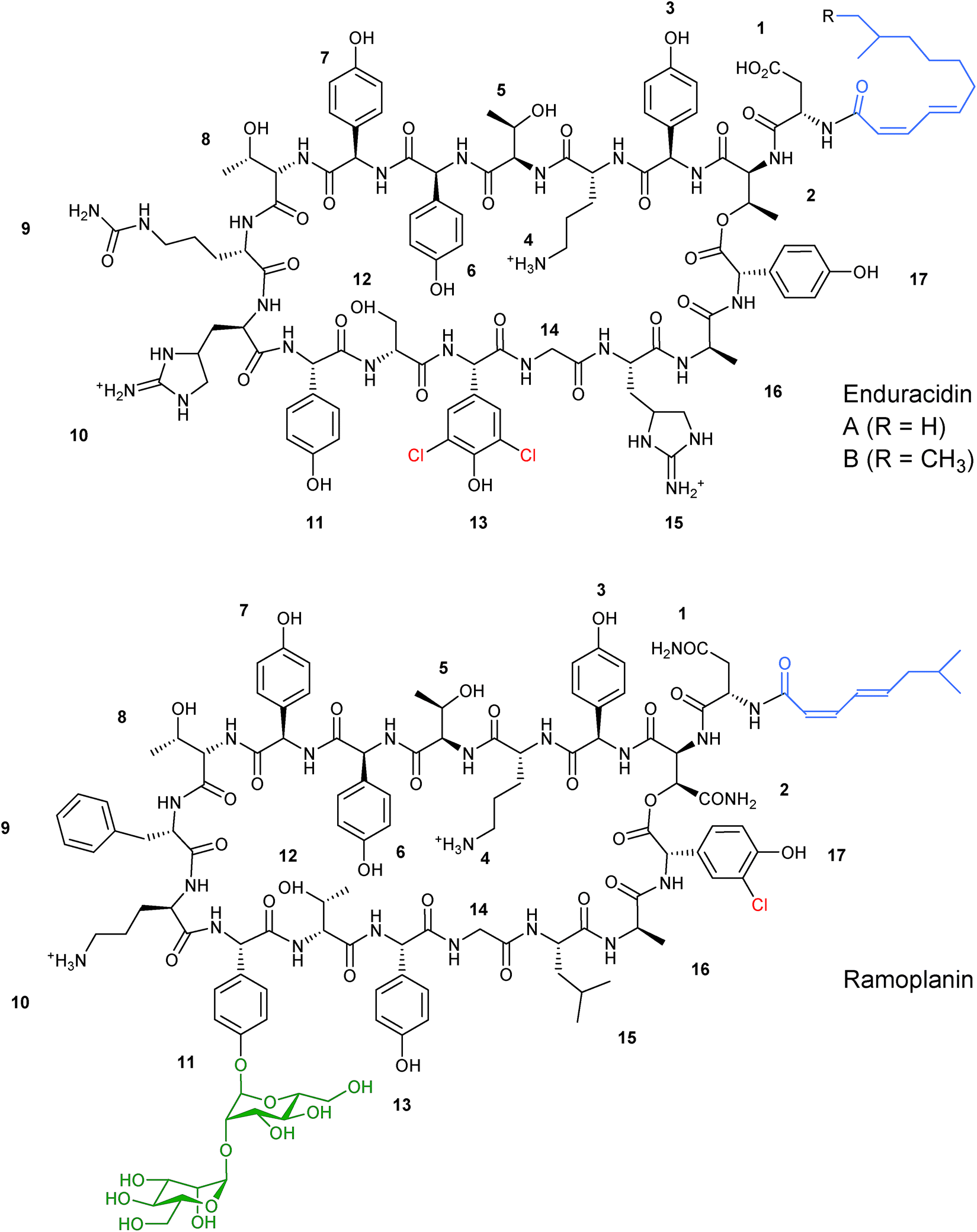 | ||
| Fig. 8 Structures of related lipopeptides enduracidin and ramoplanin. The two structures differ in chlorination pattern (highlighted in red), fatty acid side chain (highlighted in blue), and glycosylation (highlighted in green).31,32 | ||
Employing the assumption that the structural similarities between the two lipopeptide structures would allow the binding and subsequent mannosylation of enduracidin, ram29 was expressed in the enduracidin-producing Streptomyces fungicidicus. An expression cassette containing the ram29 gene along with its native Shine–Dalgarno sequence under the control of the tetracycline inducible promoter and integrated at the ΦC31 site on the Streptomyces chromosome failed to produce any evidence of mannosylated enduracidin. The expression cassette was optimised by replacing the native Shine–Dalgarno sequence and GTG start codon with the corresponding sequence from the eGFP expression construct pIJ8668. This resulted in conjugates that produced novel monomannosylated enduracidins, although the new products were produced as minor products alongside the wild type enduracidin. The site of mannosylation was determined by tandem mass spectrometry to be on L-Hpg11, the same as found in ramoplanin. The failure of enduracidin to be mannosylated twice, as with ramoplanin, is unexplained at this time, however the authors hypothesised that another enzyme outside of the ramoplanin cluster could be conducting this second mannosylation in ramoplanin biosynthesis or that S. fungicidicus could contain an α-mannosidase that may be removing one of the mannosyl groups.38 This work successfully highlighted that expression of PPM-dependent glycosyltransferases could be used as a method to produce novel glycopeptides. In addition, the importance of expression cassette optimisation when engineering natural product clusters is worth noting.
Other lipopeptides also utilise glycosylation to modulate their activity. Teicoplanin A2-2 and the related A40926 are lipoglycopeptide antibiotics used as last-line treatments for multi-drug resistant Gram-positive bacterial infections.39,40 Both lipoglycopeptides have glucosamine derived glycosyl groups, with a long N-acyl side chain (Fig. 9). This acyl chain is vital for activity and is derived from the corresponding acyl-CoA thioester by an N-acyltransferase (NAT) enzyme present in both clusters. Sequence analysis of these NAT enzymes suggested they have a unique structure not found previously.41 Syue-Yi Lyu and colleagues solved the crystal structures of these unusual NAT enzymes and found some unique traits that suggested that they represent a new NAT architecture. Based on the crystal data, in combination with biochemical and mutagenic assays, they proposed that acyl-CoA first binds to the enzyme, triggering a conformational change which forms the teicoplanin psuedo-aglycone binding site. Following the acyl transfer, the departure of CoA enables the enzyme to re-adopt the open conformation and release the acylated antibiotic. The structural information highlighted that the acyl chain extends into a spacious tunnel. The authors found that this pocket could accept a variety of long and bulky acyl chains including stearoyl (29), biphenylacetyl (36), or naphthaleneacetyl (37), and allowed the generation of a series of new glycopeptide analogues. Steric limitations prevented the acceptance of branched chains such as benzoyl-, malonyl- or methylmalonyl-CoA and ITC analysis showed that C10, the naturally incorporated chain length, was the optimal chain length for the enzyme with efficiencies decreasing as chain length was lengthened or shortened. However these results suggest that chain lengths longer than 16 may also be well tolerated, postulated to be due to the longer lipid chain forming a new favourable shape in the active site (Fig. 10). In addition to a range of monoacylated products, diacylated compounds were also formed including a 2-N-decanoyl-6-O-octanoyl-teicoplanin (43) (Fig. 11). The authors were able to test a number of these new compounds with variable length acyl chains for activity against known vancomycin resistant enterococcus (VRE) and revealed some very encouraging biological activities. In particular diacyl analogues showed significantly enhanced bactericidal activity against the tested strains when compared to mono-N-acylated teicoplanin.
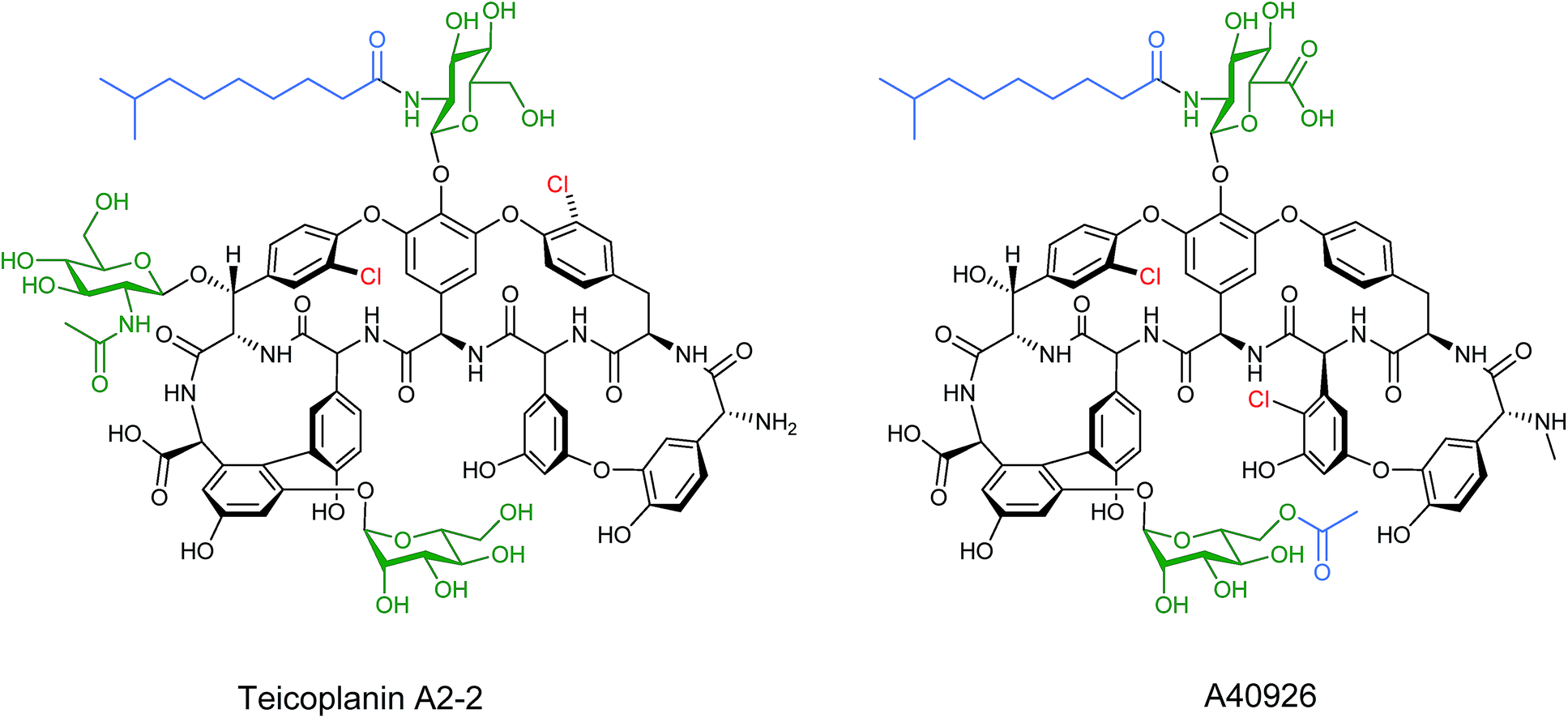 | ||
| Fig. 9 Structures of teicoplanin A2-2 and the related A40926. Glycosylation sites heighted in green and acylation sites in blue.39,40 | ||
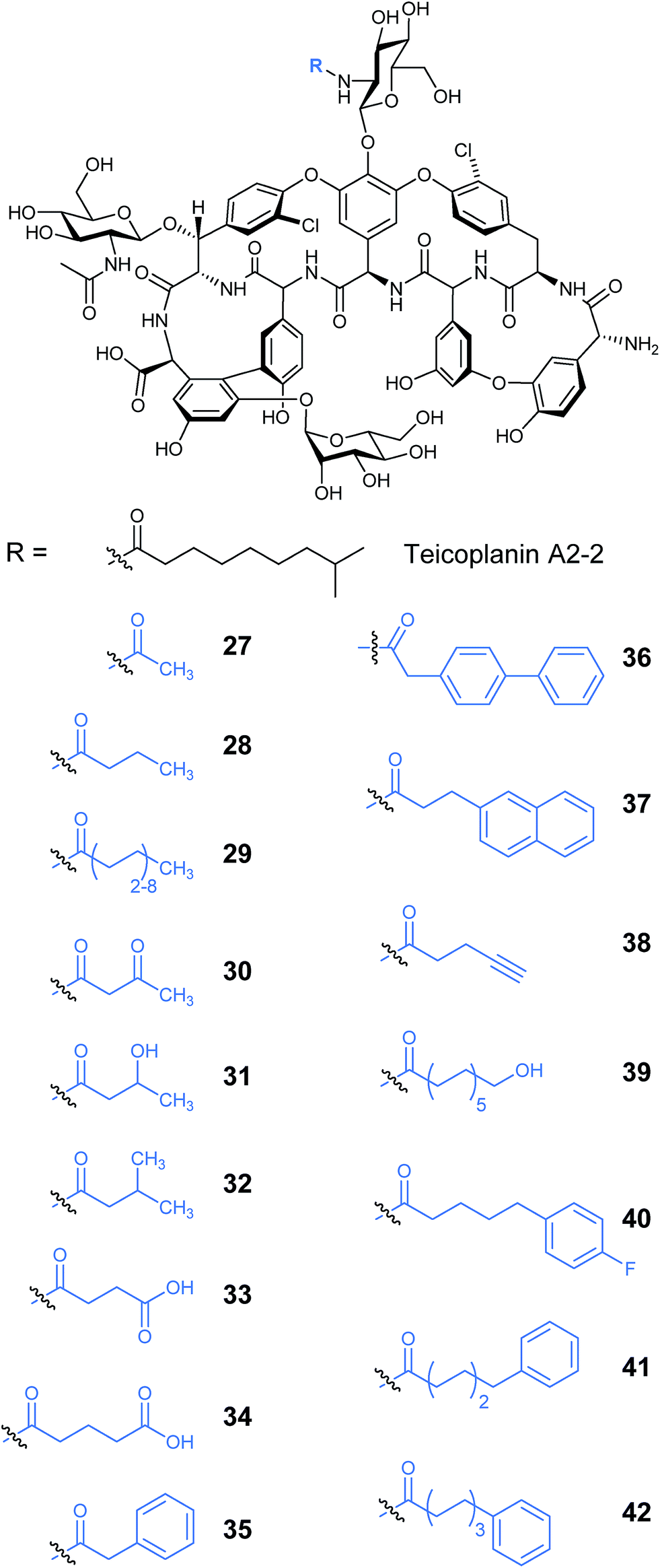 | ||
| Fig. 10 Produced mono-acylated teicoplanin A2-2 analogues.41 | ||
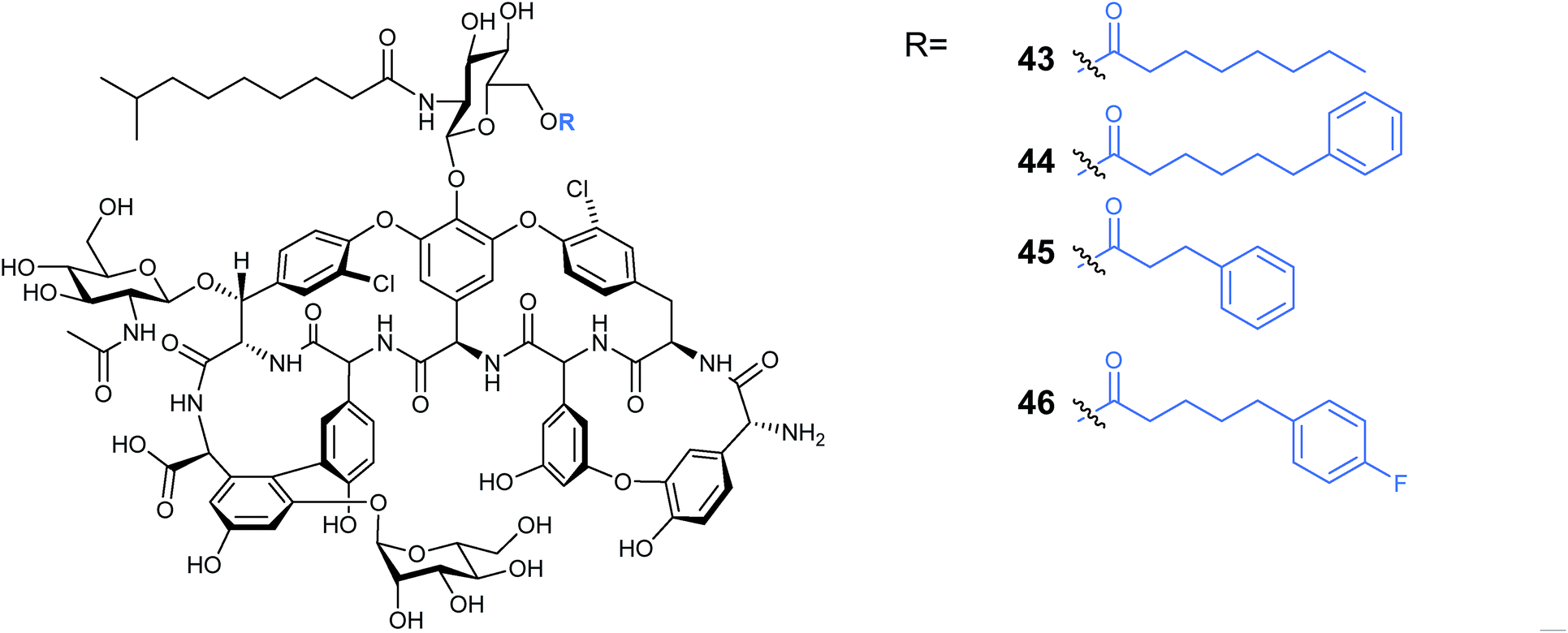 | ||
| Fig. 11 Produced di-acylated teicoplanin41 analogues. | ||
Recently another potential diversification option has been exploited which relies on the use of 3′-phosphoadenosine 5′-phosphosulfate (PAPS)-dependent sulfotransferase enzymes to modify teicoplanin-like antibiotic scaffolds. Noteably, two glycopeptide clusters were identified from an environmental DNA library (eDNA) extracted directly from soil.42 It was discovered that one of these clusters, the teicoplanin-like eDNA derived gene cluster (TEG), included several unique sulfotransferase-like enzymes (TEG12, 13 and 14). These three enzymes were heterologously expressed in E. coli and IMAC purified in order to test their activities in vitro. The nonribosomal peptide product of the TEG cluster was predicted to be very similar to teicoplanin, with the only difference being the substitution of the tyrosine found in teicoplanin to the β-hydroxytyrosine (Bht2) found in TEG. The teicoplanin aglycone (47) was therefore tested as a surrogate substrate for the three enzymes in the presence of PAPS. Each of the three TEG sulfotransferases produced a monosulfated analogue of teicoplanin (48–50) and when all three enzymes were used in tandem a trisulfated product was formed (54) (Fig. 12), suggesting that each sulfotransferase has a particular regioselectivity with TEG12, 13 and 14 sulfating the hydroxyls on Hpg3, Cl-Bht6 and Hpg4 respectively. Although these enzymes were not tested in vivo they demonstrated a potential new class of important tailoring enzymes. The related sulfonated peptide A47934 which had been isolated previously,43 was shown to be a weaker inducer of GPA resistance genes in actinomycetes compared with the corresponding desulfo-derivative.44 Based on this it was suggested that sulfation, which does not compromise anti-microbial activity, could be utilised to evade resistance to this class of antibiotics.
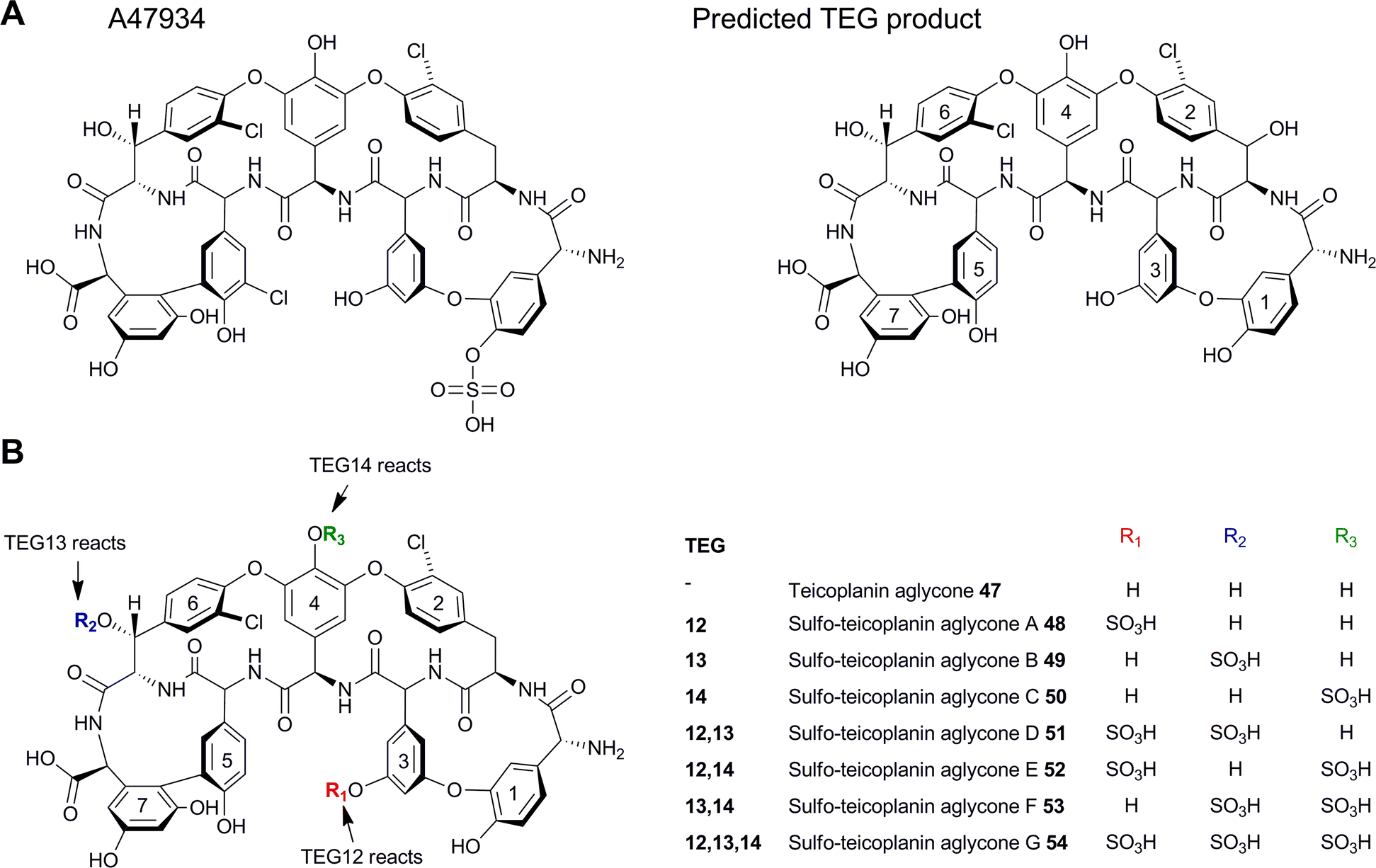 | ||
| Fig. 12 (A) Structures of sulfated teicoplanin-related compound A47934 and the predicted product from the TEG pathway. (B) Structures of teicoplanin aglycones modified with TEG sulfotransferases. TEG12, 13 and 14 reaction sites are highlighted.42 | ||
During the characterisation of the 81-kb gene cluster involved in the biosynthesis of the unusual sulfated glycopeptide antibiotic UK-68597 from Actinoplanes sp. ATCC 53533,45 a number of potentially interesting tailoring enzymes were identified that are responsible for installing a number of features on UK-68597,46 including an aryl sulfate ester on Dpg3 (dihydroxyphenylglycine), four aromatic chlorinations and an α-keto acid in place of an amino acid at the N-terminus (Fig. 13). Even though in this study UK-68597 could not be detected following fermentation, the putative enzymes involved in its biosynthesis were assigned from genome sequencing analysis. In particular, the enzyme Auk20 was assigned as a sulfotransferase, overexpressed in E. coli and then assessed for activity with various glycopeptide substrates including vancomycin, vancomycin aglycone, A47934, DS-A47934 (desulfated A47934) and teicoplanin. Both teicoplanin and DS-A47934 were successfully sulfated by Auk20 ((55) in 95% and (56) in 51% yield), with MS and NMR data placing the position of the sulfation on teicoplanin Dpg3, the same position as reported for UK-6859745 showing that the sulfation is regio-selective. The gene under the control of the ermE* promoter was also introduced into the φC31 site on the chromosome of heterologous hosts, the A47934 producer Streptomyces toyocaensis and the S. toyocaensis ΔstaL mutant, where the native sulfotransferase had been disrupted. The activity of the enzyme was monitored in cell free extracts by HPLC and MS analysis. These results confirmed the in vitro studies and showed that the desulfated DS-A47934 (produced by ΔstaL mutant) was a substrate for Auk20, producing a sulfated DS-A47934 modified at the Dpg3 moiety (56). The expression of Auk20 in the wild-type A47934 producer showed no evidence of the production of a disulfated variant (Fig. 14). Although sulfation events are rare, six different glycopeptide sulfotransferase genes have been discovered within the last 10 years. The increased rate of discovery of new sulfotransferases means that they will potentially be an important class of enzymes in the nonribosomal peptide tailoring toolkit in the years to come.
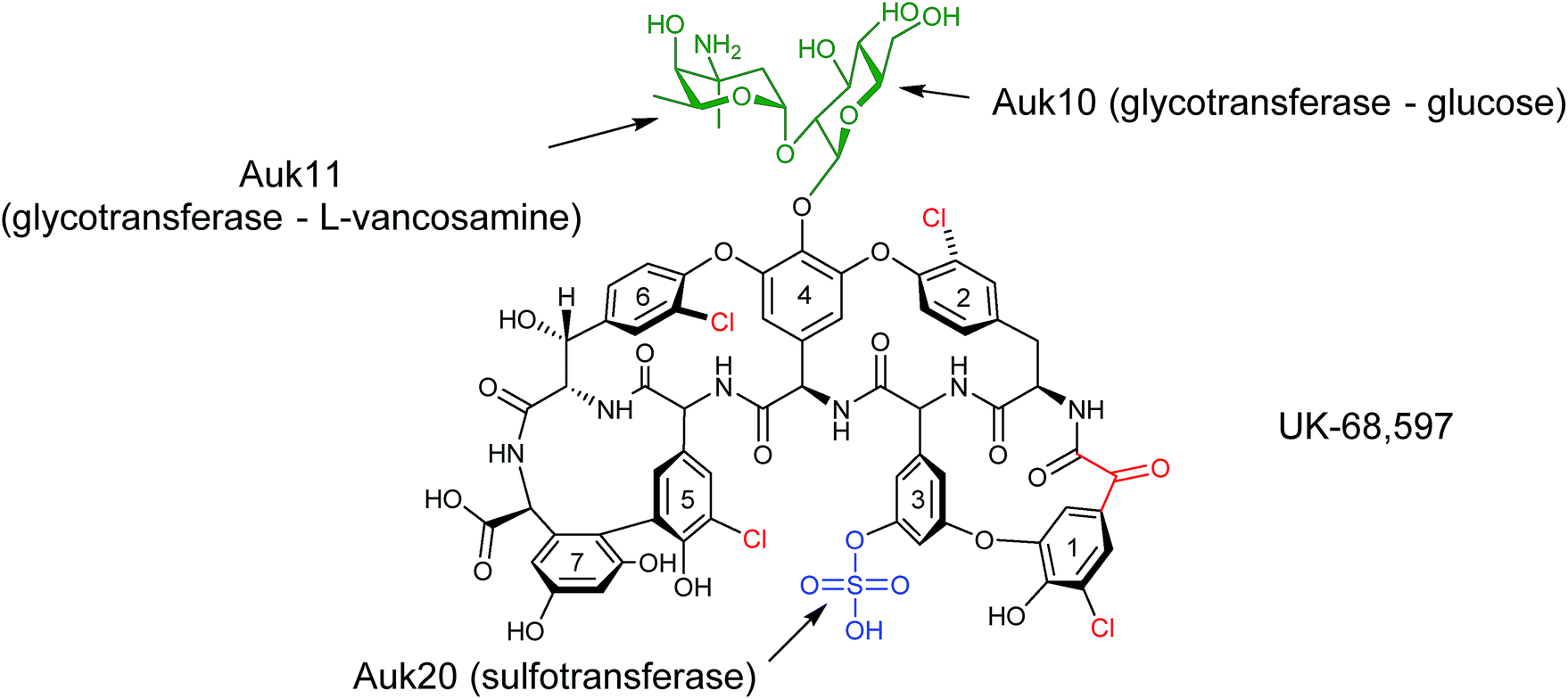 | ||
| Fig. 13 Structure of UK-68597.45 Highlighted are post-NRPS modifications around the aglycone structure (chlorination, glycosylation and sulfation). An unusual α-keto acid is moiety is also highlighted. The enzymes responsible for the sulfation and glycosylation, and sites of action, are labelled.46 | ||
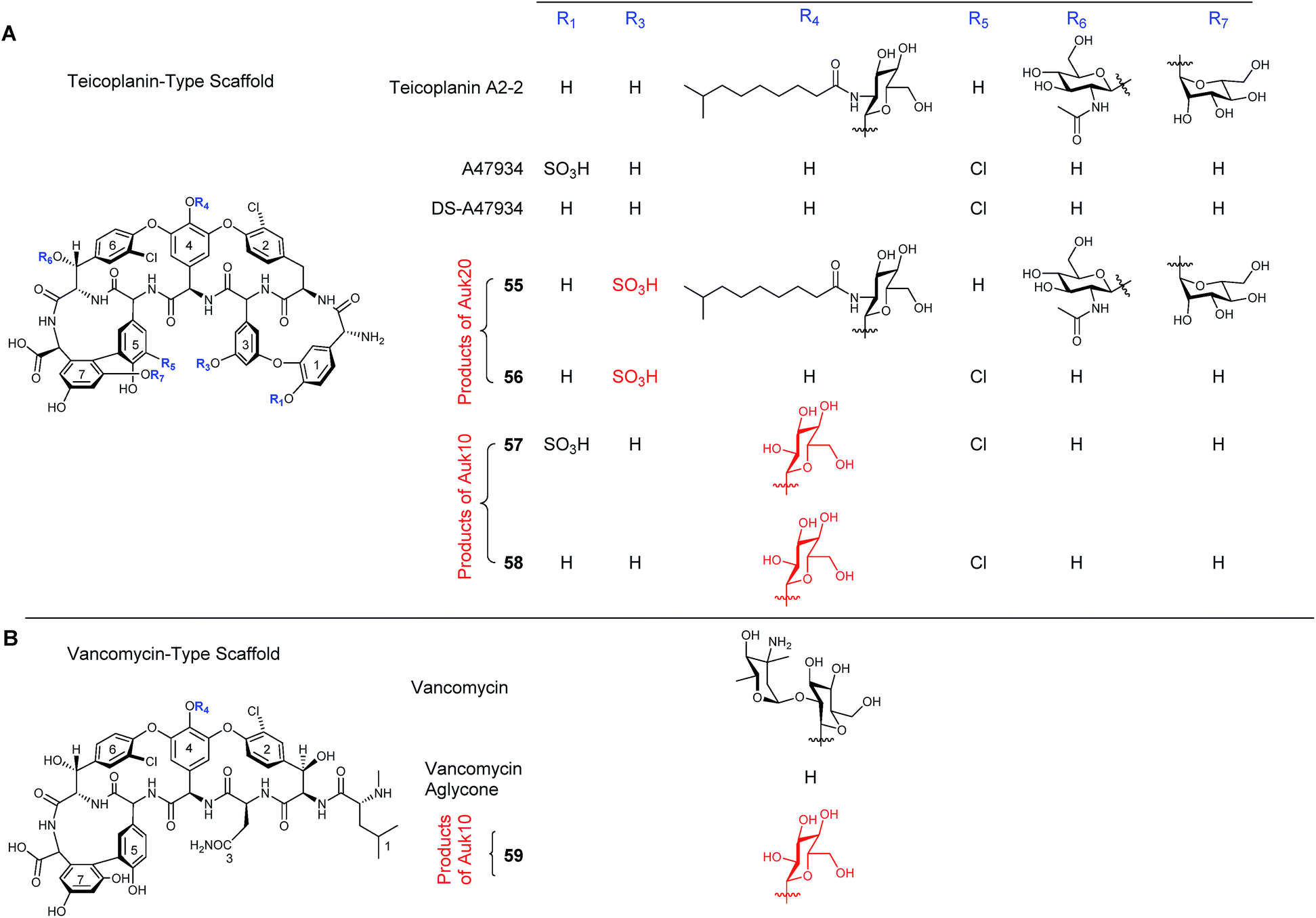 | ||
| Fig. 14 Effect of incubation of several teicoplanin- (A) and vancomycin- (B) like structures with Auk20 sulfotransferase and Auk10 glycotransferase. When incubated with Auk20 both teicoplanin and DS-A47934 showed evidence of sulfation on L-dpg3 (55 and 56). Neither vancomycin (lacking L-dpg3) nor A47934 (already sulphated at L-dpg1) acted as substrates for Auk20. Auk10 was shown to form glucosylated products from A47934 (57). DS-A47934 (58) and the vancomycin aglycone (59).46 | ||
In addition to sulfation, UK-68597 is also glycosylated with L-vancosamine-1,2-glucose at Dpg4 (Fig. 13). Three enzymes, Auk10, Auk11 and Auk14, have been identified as glycotransferases from gene cluster analysis. Auk10 showed similarity to characterised enzymes that glucosylate vancomycin on Hpg4. Auk11 showed similarities to the enzyme that installs dehydrovancosamine to balhimycin during its biosynthesis and it was, therefore, proposed that Auk10 glucosylates Hpg4 of UK-68597 while Auk11 transfers the L-vancosamine to complete the L-vancosamine-1,2-glucose glycosylation. Auk14 showed the most similarity to enzymes responsible for glycosylating amino acids at position 6 of glycopeptides such as the enzyme tGtfA that is known to install N-acetyl-glucosamine on beta-hydroxytyrosine at position 6 of teicoplanin. This enzyme, however, seems redundant as only two sugars are known to be attached to UK-68597 and none at the 6 position, although it does show some level of similarity to Auk10.
To determine which enzyme was responsible for the first glycosylation of UK-68597, both Auk10 and Auk 14 were overexpressed and purified from E. coli and tested for activity with the same glycopeptide substrates as the sulfotransferase (minus teicoplanin). Auk10 was able to glucosylate A47934 (24%) (57), DS-A47934 (8%) (58) and the vancomycin aglycone (5%) (59), while Auk14 showed almost no in vitro activity with any of the tested glycopeptides, although a trace was detected when the vancomycin aglycone was used (Fig. 14). The regioselectivity of the better performing Auk10 was determined to be position 4. As with the sulfotransferase, Auk10 was also introduced into the chromosomes of S. toyocaensis and the ΔstaL mutant but no production of glycosylated products were observed for either strain.
Together with the enzymes highlighted above, there are four additional chlorination events during UK-68597 biosynthesis and although the responsible enzymes have yet to be characterised it demonstrates that individual biosynthetic gene clusters have huge potential as rich sources of new tailoring enzymes. With the costs of genome sequencing decreasing, the number of newly characterised nonribosomal peptide gene clusters is growing which opens up the possibility that more and more unique and tantalising tailoring enzymes remain to be discovered. Discovery and characterisation of these will further increase our ability to introduce structural diversity into nonribosomal peptides during their biosynthesis. In the same environmental DNA library where the TEG pathway was discovered, a second identified cluster, the vancomycin-like eDNA derived gene cluster (VEG), was also found to encode a number of tailoring enzymes including a halogenase, 7 glycosyltransferases and 3 methyltransferases. This, in particular, highlights how environmental DNA libraries could be important to the discovery and development of new tailoring enzymes.
4 NRPS subunit, module, and domain exchanges
The above sections have shown how structural diversity can be generated by tailoring the naturally produced core peptide. An alternative, but trickier, strategy is to change the constituents of the core peptide itself. As the amino acid sequence of a nonribosomal peptide is governed by the order of the individual NRPS modules, the obvious strategy to alter this sequence is to replace or make changes within these NRPS multimeric structures. One of the problems encountered with this approach is that making even minor changes to the modular structure can effect protein folding and protein–protein interactions within the multimeric structure. Aside from the key domains themselves (C or A or T) the N- and C-terminal regions of each domain/module often act as linker regions to facilitate the association of the PCP domain with the catalytic domains. Direct module replacement can interfere with these linkers, preventing domain/module association and therefore abolishing activity. Despite these problems, however, some significant advances have been made.4.1 Exchanging NRPS subunits
Most of the key initial work done in this area was at Cubist Pharmaceuticals, a company that first rose to prominence with the development and marketing of daptomycin, a nonribosomal peptide that was the first natural product antibiotic in over thirty years to gain approval for use in the clinic.47 Daptomycin bears a high degree of structural similarity with two other lipopeptide antibiotics, namely A54145 and the calcium-dependent antibiotic. The biosynthetic gene clusters for each of these compounds produce peptides with a similar amino acid arrangement (Fig. 15), and provides an attractive starting material for combinatorial biosynthesis.47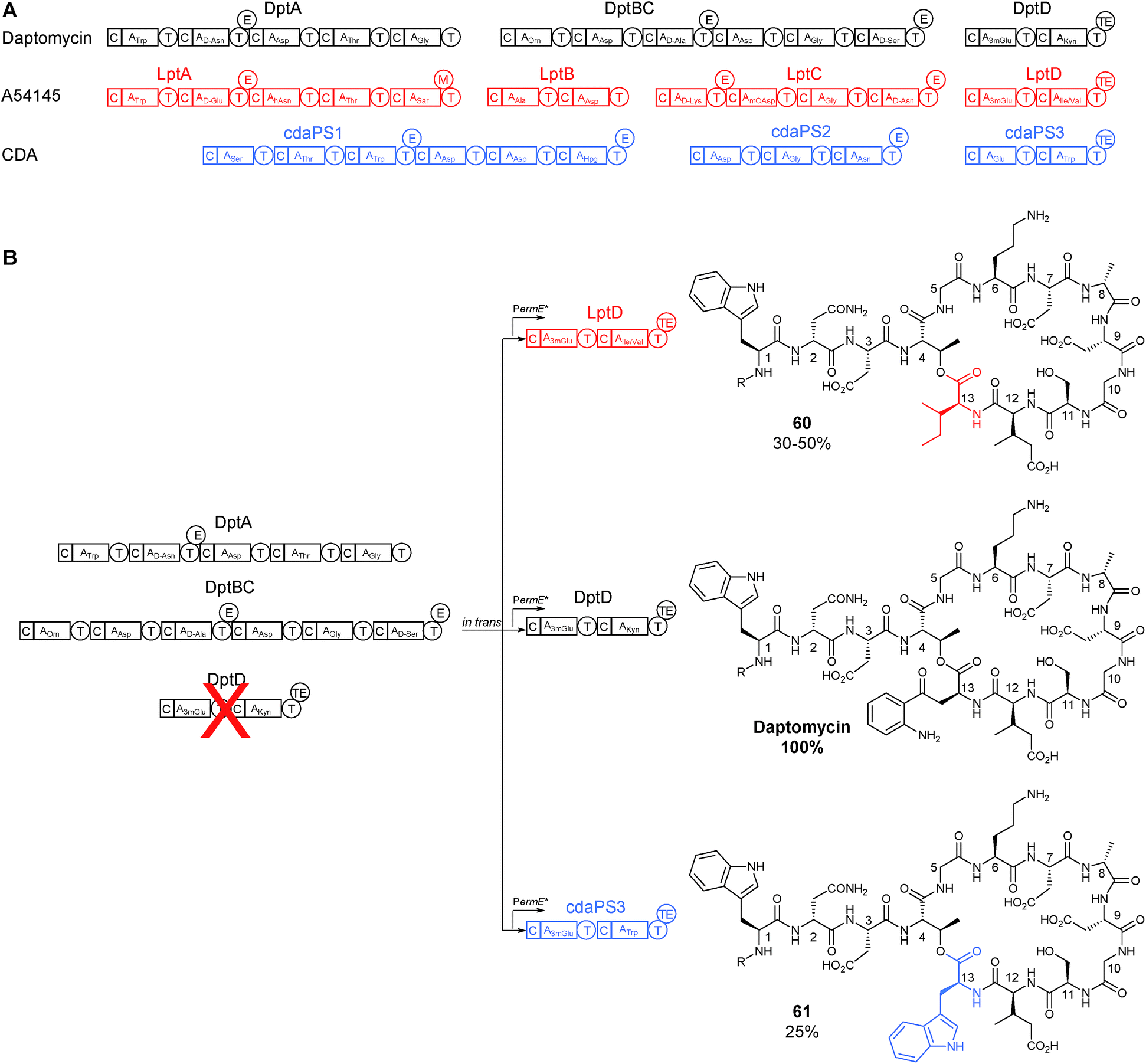 | ||
| Fig. 15 (A) Schematic of the NRPS genes responsible for the biosynthesis of the related lipopeptides daptomycin, A54145, and CDA. (B) Subunit exchange strategy where DptD subunit was first deleted and then complemented in trans with either DptD or equivalents from A54145 or CDA biosynthesis to produce two novel daptomycin analogues (60) and (61).48 R = decanoic acid. | ||
Daptomycin is a cyclic 13-amino acid lipopeptide and is a product of three biosynthetic NRPS subunits, DptA, DptBC and DptD. In the first of a series of studies, in which the biosynthetic pathway was successfully engineered to produce new derivatives of daptomycin, dptD was deleted. The gene dptD encodes for an NRPS subunit responsible for incorporating the final two amino acids, 3-methylglutamate (3mGlu) and kynurenine (Kyn), at the C-terminus (at position 12 and 13 respectively) as well as incorporating the TE domain for peptide cyclisation and release (Fig. 15).48 Following this knockout, and confirmation of the abolition of daptomycin production, successful complementation in trans was demonstrated using a strong constitutive promoter to drive expression of not only the wild-type dptD but also heterologous genes cdaPS3 and lptD from CDA and A54145 biosynthesis respectively. Both of these heterologous genes are also responsible for installing the final two amino acids in their respective NRPS pathway. CdaPS3 incorporates Glu (or 3mGlu) and Trp at the end of CDA biosynthesis, while LptD installs Glu (or 3mGlu) and either Ile or Val to finalise A54145 biosynthesis (Fig. 15). An advantage in choosing these two subunits for heterologous exchange was that they both include an initial Glu/3mGlu-specifying A domain, similar to DptD. This similarity seemed to be sufficient to maintain the interaction of the altered C domain with the upstream PCP and aided the incorporation of the subsequent non-native amino acid (either Trp, Ile or Val) at the C-terminus. Another advantage for choosing this final subunit is that the inclusion of the TE domain means that there are no downstream interactions that could be adversely affected. Although both heterologous subunit exchanges produced modified daptomycin analogues, these changes came at the expense of a drop in yield in the range of 25–50% of wild type levels (Fig. 15).48
In a follow-on study, additional genetic modifications were made in the ΔdptD strain to help to improve yields. The first module of daptomycin biosynthesis, dptA, was also deleted. This module is responsible for the initiation of the biosynthesis by first coupling the decanoic acid precursor with the N-terminal tryptophan. It was envisioned that complementing this gene in trans under the control of the strong constitutive ermE* promoter would lead to the overexpression of the initiation module and therefore positively influence yields. Using this method the production of the daptomycin derivatives was boosted to around 40–69% of wild type levels when complemented with the dptD homologous genes from either CDA or A54145 biosynthesis.49 Interestingly while all the daptomycin biosynthetic genes were seen to be expressed on a single transcript, sequential translation was not required for robust production, meaning that deletion and trans-complementation of NRPS subunits was possible.
4.2 Module and domain exchanges
The Cubist engineering approach was taken a step further when residues that make up the core cyclic peptide in daptomycin, but which are not conserved between other related lipopeptide family members, were targeted for change.50 Daptomycin contains a D-Ala residue at position 8 and a D-Ser at position 11 which are installed by modules within the second, DptBC, subunit of the daptomycin NRPS (Fig. 16B). Instead of exchanging the entire subunit for another, a smaller change was envisioned wherein individual domains (e.g. A) or modules (e.g. C–A–T or C–A–T–E) were replaced. Using λ-red-mediated recombination, the D-alanine encoding C–A–T from module 8 was deleted and replaced with the C–A–T from module 11 (which is selective for D-serine and is found downstream in the dptBC subunit). These two modules are highly homologous, making them ideal for exchange. The E domains of each module were left intact in an attempt to preserve the downstream inter-module associations. The opposite replacement was also made where the C–A–T from module 11 was replaced with the C–A–T from module 8 (change of Ser11 to Ala11). Production of the predicted D-Ser8 (67) and D-Ala11 (68) containing daptomycin analogues was observed, albeit at reduced production levels of approximately 15% and 45% relative to wild-type, but both new analogues retained activity against S. aureus.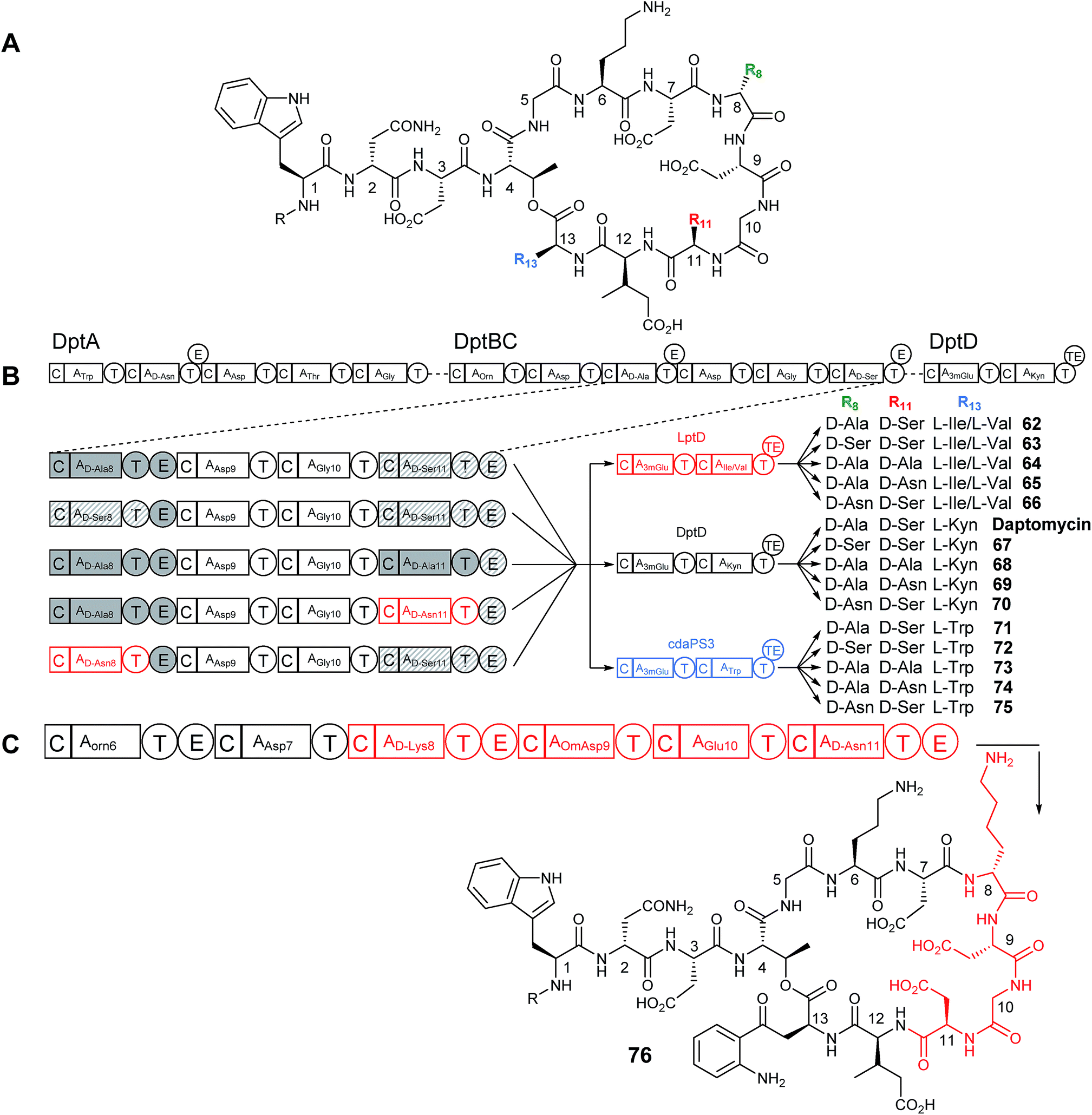 | ||
| Fig. 16 (A) Structure of daptomycin and analogues produced through NRPS module exchange. (B) NRPS organisation of the daptomycin cluster and schematic showing module exchange strategy. Modules 8 and 11 were swapped for each other or for the Asn8 module from A54145 biosynthesis (see Fig. 15A). These NRPS chimeras were then expressed alongside either the wild type DptD subunit or similar LptD/cdaPS3 subunits from A54145/CDA biosynthesis. (C) Further module swapping involved swapping an entire four subunits from DptBC for the related subunits from A54145 biosynthesis. Daptomycin modules are shown in black, A54145 modules in red and CDA modules in blue.50 Domains originating from DptBC module 8 are shaded solid gray, domains originating from DptBC module 11 are hatched. | ||
Following this proof of concept the genes from the similar A54145 biosynthetic cluster (the D-Asn encoding module 11) were used to replace either D-Ser8 or D-Ala11 positions with D-Asn (Fig. 16B). This also proved successful and two new analogues were isolated (D-Asn11 (69) and D-Asn8 (70)), however production levels were further reduced relative to wild type (the D-Asn11 (69) analogue showing slightly higher production than D-Asn8 (70)). Replacing the original E domain with the heterologous E domain from A54145 also resulted in the formation of the new analogues, demonstrating that total module replacement (C–A–T–E) is possible, but this change caused a significantly decreased yield of product versus the native E domain showing that maintaining the native module–module linker regions is important for activity.50 Activity assays showed that the D-Asn8 daptomycin analogue (70) was less active than daptomycin, but the D-Asn11 analogue (69) retained potency.
More extreme changes were also made to the core structure of daptomycin by exchanging four modules within DptBC (D-Ala8-Asp9-Gly10-D-Ser11) with four modules from LptC (D-Lys8-OmAsp9-Glu10-D-Asn11) from the A54145 cluster (Fig. 16C). Although production of the expected product of this module exchange was seen (minus the O-methylation of Asp9 due to the necessary tailoring enzymes not being present in the daptomycin cluster) (76), the yield was drastically reduced, with production being less than 0.5% of control levels.
The successful production of compounds with changes at positions 8 and 11 led to the combination of these with the previously successful changes to the final amino acids at positions 12 and 13 through exchange of dptD for lptD (62–66) or cdaPS3 (71–75) (as well as other modifications made to the lipid tail) leading to the production of multiple daptomycin hybrid compounds (Fig. 16B) with production levels ranged from approximately 0.5–45% of control levels, and a general trend observed that the greater the number of changes imposed the lower the production levels. Each compound was assessed for antibacterial activity and although they were, on the whole, no greater in potency than daptomycin (with the exception of one against an E. coli imp mutant), the successful production of new compounds in a combinatorial manner indicates that this sort of approach is possible.
In a follow on study dptD was again chosen to be the subject of modification. The C–A or C–A–T domains of module 13 (incorporates Kyn13 in daptomycin) were exchanged for different domains from cdaPS3 (incorporates Trp11 in CDA) or lptD (incorporates Ile13 in A54145) (Fig. 15A). Although no production was observed in the C–A–T domain swaps, exchange in just the C–A domains alone led to production of the predicted Trp13 and IIe13 daptomycin analogues at levels of approximately 20% of the control. Unfortunately the new compounds produced were found to be less potent than daptomyin when assessed in antibacterial assays.51 The fact that Cubist failed to identify any daptomycin variants with improved antimicrobial activity, despite a major industrial effort, is perhaps not surprising. There is a very close evolutionary relationship between the daptomycin, CDA & A54145 NRPS modules and domains that were exchanged. Most likely nature had already sampled these combinations and selected against the compounds Cubist created, in favour of the daptomycin which remains the most active antibiotic in this large family of natural and engineered lipopeptide variants.
A slightly more subtle domain swapping approach was demonstrated with the in vivo production of novel pyoverdine derivatives by Pseudomonas aeruginosa PAO1 through smaller domain substitutions where alterations were limited to either the A domain alone (A) or together with the C domain (C–A).52 Similar to dptD, pvdD encodes an NRPS subunit responsible for incorporating the final two residues, which are both threonine at positions 10 and 11 of pyoverdine (Fig. 17A, B). A previous attempt where changes were aimed at altering the penultimate amino acid in pyoverdine had failed, presumably due to disruptions in the interactions between modules in the system.15 In an attempt to minimise these disruptions only the final amino acid in pyoverdine (Thr11) was substituted with alternative amino acids. In a similar fashion to the earlier daptomyin experiments, the native A domain (or both the A and C domains) of module 11 of pvdD was first deleted and then expressed in trans to ensure successful complementation was possible before various A domain (or C–A domains) replacements were introduced. Replacement domains tested included homologous domains obtained from elsewhere in the same biosynthetic cluster (Fig. 17B), or heterologous genes obtained from pyoverdine biosynthetic clusters present in other Pseudomonas species (Fig. 17C). In total, nine constructs for each of the A and C–A domain replacements were complemented into the ΔpvdD strain; three Thr-specifying, three Ser-specifying (one of which accepted a Thr from the neighbouring module immediately upstream), one Lys-specifying, one Asp-specifying and one Gly-specifying. Each of these were assessed for production of pyoverdines, detected either by monitoring changes in UV or a more sensitive fluorescence method.
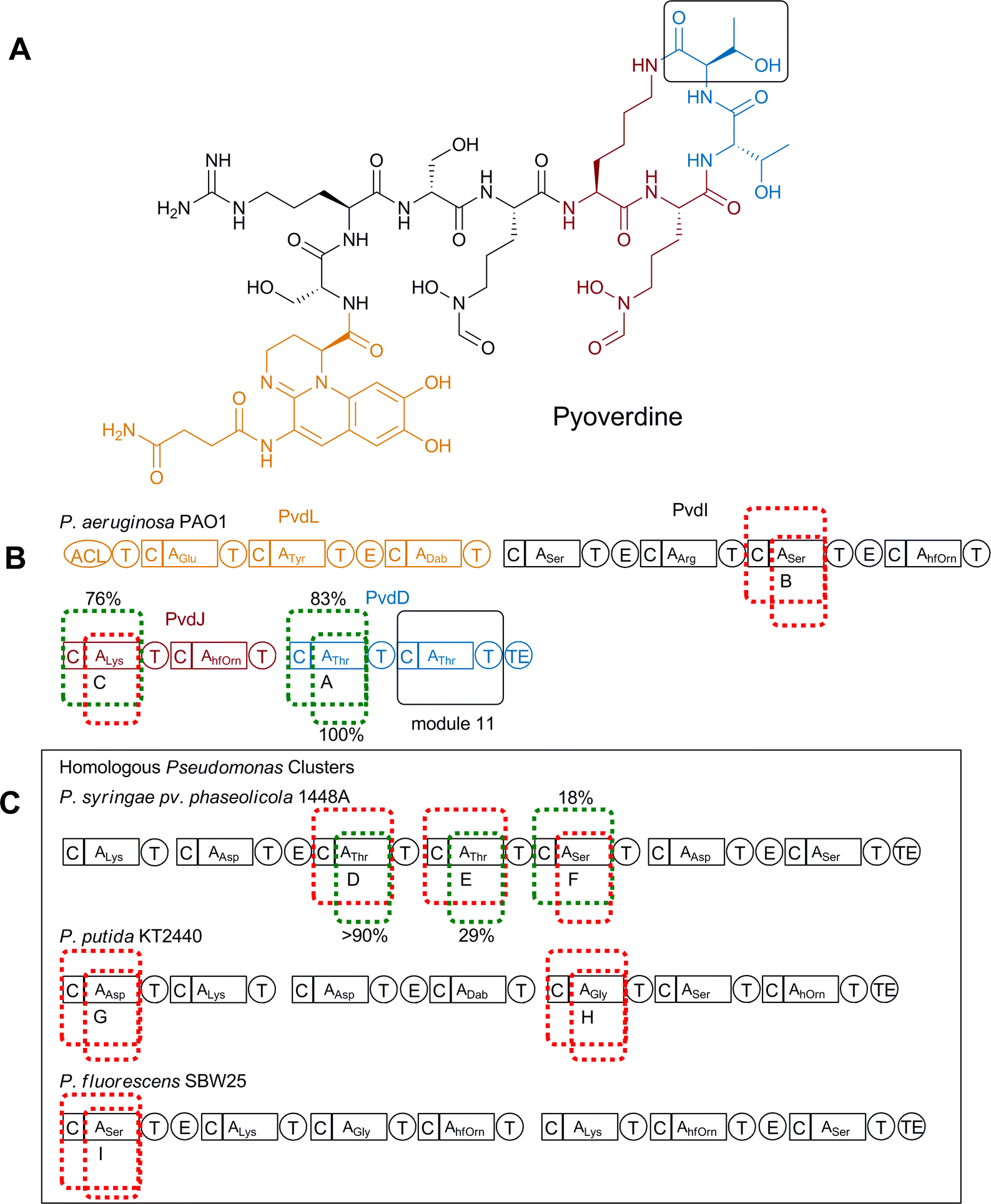 | ||
| Fig. 17 (A) Structure of pyoverdine. Amino acid chosen for replacement is shown in box. (B) Module replacement strategy for the replacement of Thr11 (shown in black box). Module 11 was replaced with three modules from within the pyoverdine cluster. Replacement modules (either C–A or just A) are highlighted with dotted lines. Modules that were successfully complemented are shown in green, unsuccessful in red. (C) Modules from homologous Pseudomonas clusters that were used to replace PvdD11 are highlighted within their native clusters. Modules that were successfully complemented are shown in green, unsuccessful in red. Relative production levels of successful chimeras are shown.52 hfOrn = L-N5-formyl-N5-hydroxyornithine. | ||
Where the native Thr11 A domain was substituted with the three non-native Thr-specifying A domains production of pyoverdine could be detected by absorbance in each case, with two showing native production levels and the third producing at 29% of the control level. All the other, non-Thr-specifying A domain substitutions, were found to produce very low levels of a pyoverdine-like compound that could only be detected using the more sensitive fluorescence detection. Mass spectrometric analysis showed that all these analogues still contained Thr11, indicating that these heterologous A domain replacements had failed to function as anticipated in their new context.
Differing results were obtained with replacement of the C–A domains, with only one of the homologous Thr-specifying replacements (from the P. syringae pyoverdine biosynthetic gene cluster) producing high levels of pyoverdine (83% of the control). This result taken in isolation would seem to indicate that joint C–A domain replacement was a less favourable approach than the simpler A domain replacement, however, unlike with isolated A domain replacement, two novel pyoverdine derivatives with unnatural substitutions at position 11 were successfully obtained when replaced with non-threonine C–A substitutions. One of these, containing Lys11, was obtained at 76% of control level and was produced following domain exchange with a C–A domain taken from pvdJ from the same pathway in P. aeruginosa PAO1. The second, containing Ser11, was obtained at 18% of control level and was produced following replacement with a C–A domain from P. syringae pv. phaseolicola 1448A (which in its native context accepts Thr from an upstream neighbouring module). It was also reported that in the majority of C–A domain replacements a truncated product related to pyoverdine was detected, indicating stalling of biosynthesis as a result of the modifications. This study highlights that a single approach to engineering NRPS specificity is not always applicable and that domain substitutions do not always function in a predictable fashion. It also highlights that the condensation domains (C) may be performing a more complicated role than just peptide bond formation and may have some role in substrate selection.
4.3 Module deletions and insertions
In all of the studies described above, the main focus was to alter the residues incorporated at a given position within the nonribosomal peptide, either individually or in combination, to create novel structures. Another route that has been explored is to alter the length of the peptide chain. Modifications that have been explored include module deletion, which has previously been demonstrated to result in reduction in the ring size of surfactin,53 or insertion of one or more modules to expand the ring size. For example, an additional amino acid was inserted into a central position of balhimycin, a glycopeptide antibiotic composed of seven residues. To achieve this a new module was inserted between the fourth and fifth modules of the balhimycin NRPS biosynthetic gene bpsB. This new module was a chimera of modules 4 and 5, both of which add an D-Hpg to the elongating peptide. In an attempt to maintain correct module–module interactions, the new module was composed of the C–A domains of the fifth module, combined with the T and E domains taken from the fourth module. This approach led to production of an octapeptide containing three consecutive D-Hpg residues. However, a number of truncated products were also obtained as well as products lacking cyclisation and further modifications,54 suggesting some incompatibility of the new product with downstream processes.It was concluded that the relationships between the modules upstream and downstream of inserted domains were important to successfully create hybrid NRPS assembly lines. Whilst the C–A domain from module 5 was kept intact, as this was deemed important for conferring D-Hpg specificity, the arrangement of the other domains were aligned so that the native linker regions were maintained. So while the C–A of module 5 was chosen to maintain connection with the upstream E of module 4, in the same way the epimerisation domain (E) from module 4 was chosen to make the most efficient contact with the downstream C domain of module 5.54
4.4 Importance of module–module linker regions
In order to produce a successful NRPS domain alteration it is important to pay careful consideration to the interfaces between the native and non-native domains. For example, work on daptomycin domain exchange showed that mutation, insertion or deletion of up to four amino acids in the T to C linker region (between neighbouring units) had no deleterious effects on production of daptomycin. Similarly, alterations in the domain linker regions within module 13 were well tolerated when the native Kyn was being incorporated at position 13, however when the Kyn activating C–A–T domain was exchanged for a Asn activating domain, production was only observed when the native T to TE domain linkage was maintained indicating that, at least under some circumstances, the T to TE linkage should be considered important to correct operation of the NRPS.51In another example, during dissection and heterologous expression of the three module beauvericin and bassianolide biosynthetic systems it was shown that product formation relied on maintenance of the N-terminal linker region of the C domain of the second module.55
Another recent study has looked into the effect of substituting the native T domain of IndC, from the indigoidine biosynthetic pathway of Photorhabdus luminescens, with a number of heterologous synthetic T domains selected following a computational analysis. In total seven synthetic T domains were assessed that showed either high homology, less homology or little homology to the native one and it was found that one third of the synthetic systems were functional. In addition the T domain from BpsA, a IndC homolog from Streptomyces lavendulae, was also tested. Due to the similarity of BpsA it was expected to yield a functional enzyme, however it proved to be nonfunctional. The problem was thought to be caused by poor inter-domain interaction, therefore a number of genetic constructs with different A to T and T to TE linker regions of IndC or BpsA origin were produced and assessed. It was discovered that inclusion of longer lengths of the native linker regions originating from the incoming BpsA positively affected indigoidine production, indicating again that native linker regions are often essential for correct NRPS activity.56
Further importance of the linker hinge regions, in this instance between A and T domains, was shown in studies on EntF, an NRPS involved in enterobactin biosynthesis. A D857P mutation was introduced into the A–T linker region, this was based on previous work on acyl-CoA synthetases which showed that insertion of a proline in the hinge region restricts subdomain rotation and traps the enzyme in the adenylate forming conformation.57,58 As expected, this mutation in EntF abolished production of enterobactin in a reconstitution assay, despite detection of wild type levels of adenylation activity in a PPi exchange assay. This confirmed that the hinge region conformation, which the proline interferes with, is important for domain alignment and catalysis. Interestingly, subsequent sequence analysis of multiple A–T domains then revealed that the region following the A10 motif (the Stachelhaus A domain specificity conferring residues)4,5 is more proline-rich than those found in standalone A domains. Mutation of one (P961) or a combination of other proline residues (P959, P968 and P972) led to severely impaired production of enterobactin. A conserved LPxP motif was then identified at the N-terminus of the A–T domain linker region and shown, through homology modelling and further mutational analysis, to interact with a key residue (Y908) in the C-terminus of the A domain which is also required for movement of the T domain relative to the A domain to complete the catalytic cycle.59 This suggested that the linker regions need to form a specific conformation for activity which is in part controlled by these proline residues. This in vitro study provided mechanistic insight and biochemical evidence of the importance of linker regions in controlling domain conformation and lends greater weight to previous observations that suggest careful consideration of these regions should be undertaken when attempting any combinatorial biosynthesis studies with an NRPS.
4.5 Exchanging of sub-domains
The complicated relationship between NRPS domains and linker regions has led to the development of a new tactic where only an internal sub-domain, which has no direct contact with other modules, is exchanged. This has allowed changes to be made to an adenylation domain without affecting the structure of the native linker regions. The approach was developed based on insights gained into the evolutionary pathway of the NRPS genes involved in hormaomycin biosynthesis. It is thought that a natural recombination event occurred during the evolution of the NRPS genes of this biosynthetic pathway which greatly altered the substrate specificity of the adenylation domain. Crüsemann et al. examined the sites of this natural A domain exchange, and performed homology modelling to ensure that these were not in regions that would adversely affect secondary structure. These natural recombination sites were then used as guidelines to direct their mutations.The identified core sub-domain of the HrmO-(β-Me)Phe3 A domain (Fig. 18A & C) was replaced with three different sub-domains taken from other NRPSs in the hormaomycin biosynthetic pathway (HrmO-(3-Nep)Ala4, HrmO-Thr2 and HrmP-Val6) as well two sub-domains from CDA biosynthesis (cdaPS1-Asp5 and cdaPS1-Hpg6). The hybrid domains were then expressed and purified for use in adenylation activity assays. Although the hybrids derived from the sub-domains of the hormaomycin pathway were all active and recognised their cognate amino acid, those derived from the CDA biosynthetic pathway were inactive.60
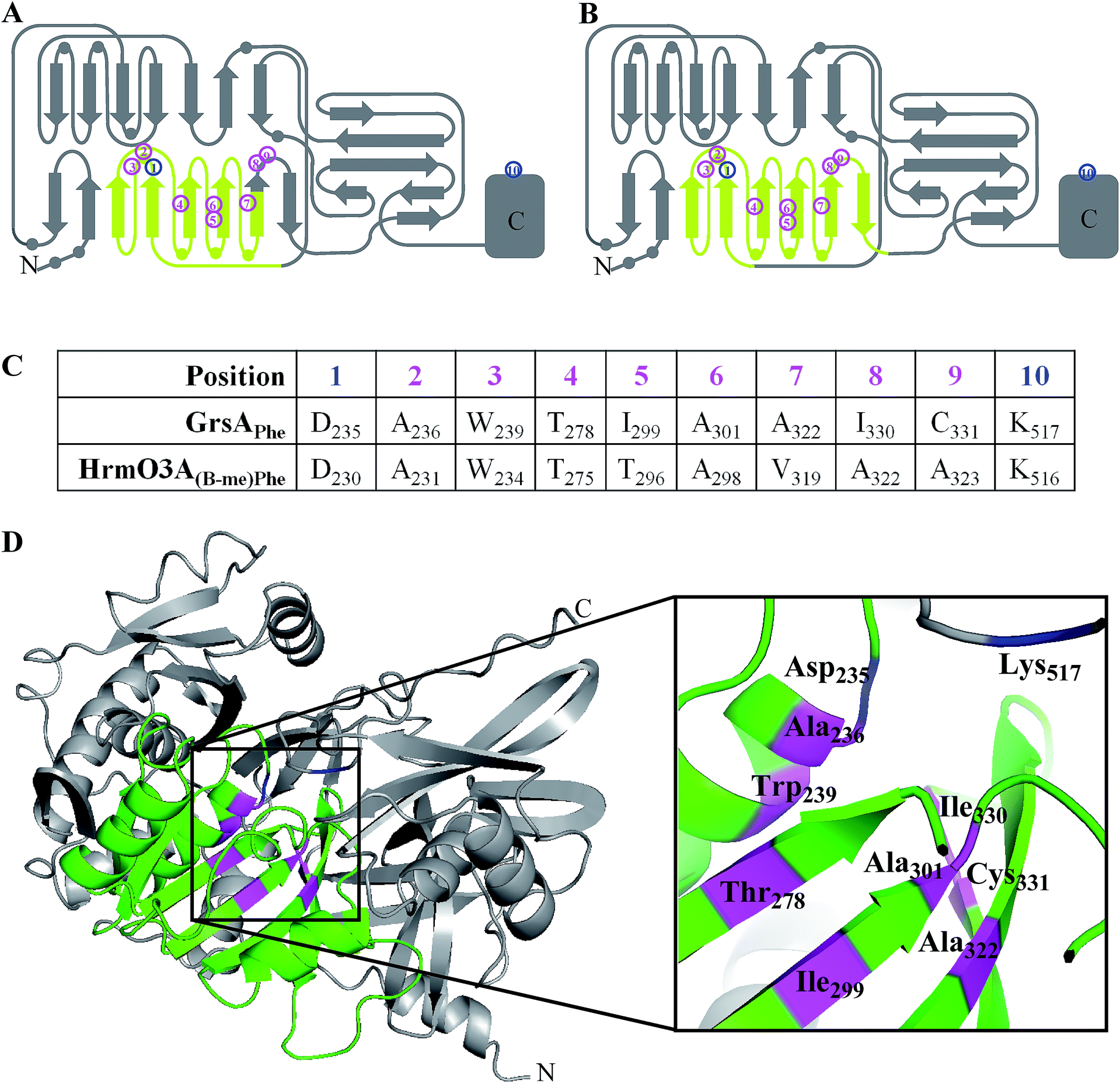 | ||
| Fig. 18 (A) & (B) 2D representation of adenylation domain secondary structures with circles representing helices and arrows representing sheets (adapted from Kries 2015).61 The swapped sub-domains are highlighted in pale green spanning residues 204 to 323 (A);60 or residues 221 to 352 (B).61 Magenta numbers highlight the eight substrate specifying residues and blue numbers highlight the invariable Asp and Lys residues. (C) The specificity-conferring codes of HrmO3A and GrsA. (D) 3D structure of the Phe-activating adenylation domain of GrsA3 with the flavodoxin-like domain coloured green. The eight substrate specifying residues are highlighted in magenta and the invariable Asp and Lys residues are highlighted in blue. | ||
Subsequent work by a different group identified further boundaries for replacement that were limited to only the flavodoxin-like sub-domain of the Phe-specifying A domain of GrsA (Fig. 18B and D). Nine sub-domain replacements were made, four taken from other NRPS subunits in the gramicidin biosynthetic pathway, and five from NRPS subunits of a range of other biosynthetic pathways. The hybrid domains were then expressed and purified for use in adenylation activity assays. Significant adenylation activity was shown for two sub-domains taken from the gramicidin pathway as well as two from other species, showing that the success of sub-domain transplantation is not restricted to within a particular species.61 A further assay coupled the activity of the hybrid A domain mutants to GrsB1 in an attempt to produce a diketopiperazine. In this case, one out of the four mutants that had previously been identified as successful via the A domain assay was tested and shown to catalyse this reaction. The fact that even one out of the nine flavodoxin-like sub-domain mutants was successful in both of the in vitro biochemical assays demonstrates the ability of this approach to form a functional A domain which, with further refinement, could be extended for use in other biosynthetic pathways and for in vivo production of hybrid nonribosomal peptides.
4.6 Perspective and future of NRPS subunit/module/domain exchanges
From the work presented here it is clear that more often than not, when taken out of their native context, NRPS domains fail to function in an optimal manner. Whether this takes the form of an inability to recognise and/or activate their substrate, an inability to incorporate it into the growing nonribosomal peptide during synthesis or through stalling of synthesis post-incorporation it ultimately negatively impacts the production of the desired final nonribosomal peptide product. It is interesting to see that expression of some synthetic T domains, designed with high homology to the native T domain, actually led to enhanced production of indigoidine.56 Computer-aided modelling of indigoidine production suggests that this may be due to a reduced presence of a toxic precursor being available in this mutant due to an increased rate of dimerisation of this precursor by the mutant enzyme. Rational design of biosynthetic pathways may, therefore, also provide a means to ensure that changes in an NRPS module, domain or subdomain do not have deleterious effects on production. However significant work still needs to be done to fully understand the process of domain swapping to enable true combinatorial biosynthesis of the NRPS scaffold.5 Active site modification and directed evolution of adenylation domains
The adenylation domain specificity code that specifies which amino acid is incorporated into a growing peptide has been deciphered.4,5 Changes to individual amino acids within a protein structure are, in general, less likely to cause major permutations to the overall structure and, in the case of NRPS domains, are less likely to introduce disruptions in the inter-domain linking regions. Through the introduction of individual or combined point mutations in the binding pocket of an NRPS adenylation domain, incorporation of a non-native amino acid into the nonribosomal peptide can be achieved or in the case of promiscuous adenylation domains, the production profile can be altered so that the desired product is the major or only product of the system.3,45.1 Changing selectivity for alternative natural amino acids
An example of a promiscuous adenylation domain is the third module of the NRPS FusA, which is responsible for the biosynthesis of the fusaricidins. Fusaricidins have a hexapeptide core and FusA is known to be able to incorporate either L-Tyr, L-Val, L-Ile, L-(allo)-Ile, or L-Phe as the third amino acid. The fusaricidins are therefore produced as a mixture of a number of related compounds.63 Fusaricidins have bioactivity against both fungi and Gram-positive bacteria with the L-Phe-containing variant showing the best antibiotic activity compared to those containing alternative amino acids at the 3 position.63,64 With a view to driving biosynthesis preferentially towards production of this active analogue, the substrate binding pocket of the third adenylation domain in FusA was aligned with the known Phe-specifying A domains GrsA and TycA (Table 1). From this comparison it was hypothesised that the mutation of four residues (S239W, L299I, G322A, and V330I) would shift the selectivity of FusA towards Phe.65 Six different constructs were prepared of FusA which contained one or more of these mutations. Following fermentation of strains containing these constructs, preferential production of the Phe-containing analogue was seen in three out of the six mutants (Table 1). When all four residues were altered to those found in GrsA and TycA, production of the Phe-containing fusaricidin analogue was three times higher than wild-type. Two other mutants harbouring double and triple mutations also produced elevated levels of the Phe-containing fusaricidin analogue (2.5 and 2 times higher compared to wild-type). Despite all three high-producing mutants harbouring the L299I mutation, this alone was not shown to be sufficient to increase production levels of the Phe-containing analogue, indicating that the effects of the mutations may be cumulative.
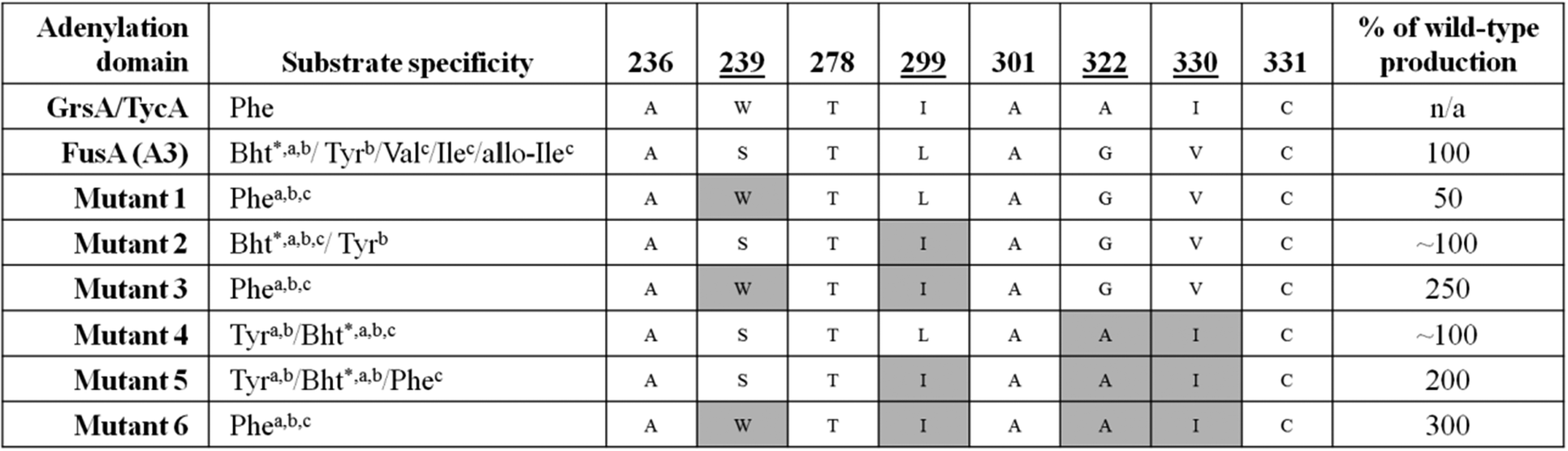
|
Another study examined the third module of Plu3262, from the luminmide NRPS, which includes a promiscuous adenylation domain that incorporates predominantly Phe (producing luminmide A), or Leu (producing luminmide B). Expression of the luminmide biosynthetic genes, taken from an entomopathogen, in E. coli allowed luminmide production to be examined in a non-pathogenic host and also led to the authors discovering that Tyr, Val or Met can also be incorporated by Plu3262 to produce novel luminmides, albeit at a much lower level. This heterologous system also permitted the use of ccdB counterselection (discussed later in this review) as an efficient and seamless means of introducing concurrent mutations into the A domain coding sequence. Based on the comparison of the specificity conferring code of Plu3262 with a range of Leu, Phe and Val-specifying adenylation domains, three residues were chosen for mutation with the aim of driving production towards luminmide B (C278M/T, I299F, and A301G). Variants of Plu3262 were produced harbouring either a single or double mutations in these residues.66 Three of these strains were found to favourably overproduce luminmide B when compared to wild-type. The best result was seen with the single mutation A301G. The two other overproducing strains also harboured this mutation but in combination with mutations that in isolation were shown to negatively influence production of luminmide B. The change in production profile between luminmides A and B was also mirrored by some of the minor luminmides, which could prove advantageous for further characterization of these newly discovered analogues.
5.2 Changing selectivity for alternative non-natural amino acids
A similar, but extended, approach allowed the incorporation of a non-natural amino acid into CDA. Module 10, contained within the cdaPS3 subunit of the CDA NRPS, normally incorporates either Glu or 3mGlu (3-methylglutamate) at position 10 of the cyclic lipopeptide. Incorporation of the non-native amino acids Gln or 3mGln (3-methylglutamine) at this position in CDA was, again, guided by multiple sequence alignments of the Glu-specifying residues in the A domain of CdaPS3 to a range of Glu and Gln-specifying A domains. These alignments suggested that Glu-activating A domains tend to possess a basic residue (Lys or His) at either position 239 or 278 and that Gln-activating A domains tended towards a Gln residue at the same positions (Fig. 19A). Based on the Lys residue being at position 278 of CdaPS3, and that other mutations may favour Gln/mGln recognition over Glu/mGlu, two mutants were made within the adenylation domain of module 10 (K278Q and Q236E). Site directed mutagenesis was used to introduce these changes into a plasmid containing the module 10 A domain, either individually or in combination, and these were then introduced via homologous recombination so that they were expressed from the native location on the chromosome of the producing organism, Streptomyces coelicolor (MT1110). Only one of the mutant strains, harbouring a single mutation (K278Q), was found to produce CDA containing Gln at position 10 (77).67 This mutation was also introduced into a ΔglmT strain, which is deficient in an enzyme required for the biosynthesis of methylated glutamate,68 thereby ensuring that any methylated CDAs observed would be due to the addition of methylated substrates to the culture. During fermentation a dipeptide of Gly-mGln was fed to the culture, intended as a source of mGln following intracellular cleavage. Although the major product of the fermentation of this mutant was the same as observed previously, production of 3mGln-containing CDA was also observed (78). As (2S,3R)-3mGlu has not been identified in nature, this work represented the first example of the introduction of a non-natural amino acid into a nonribosomal peptide through active site modification of an NRPS A domain.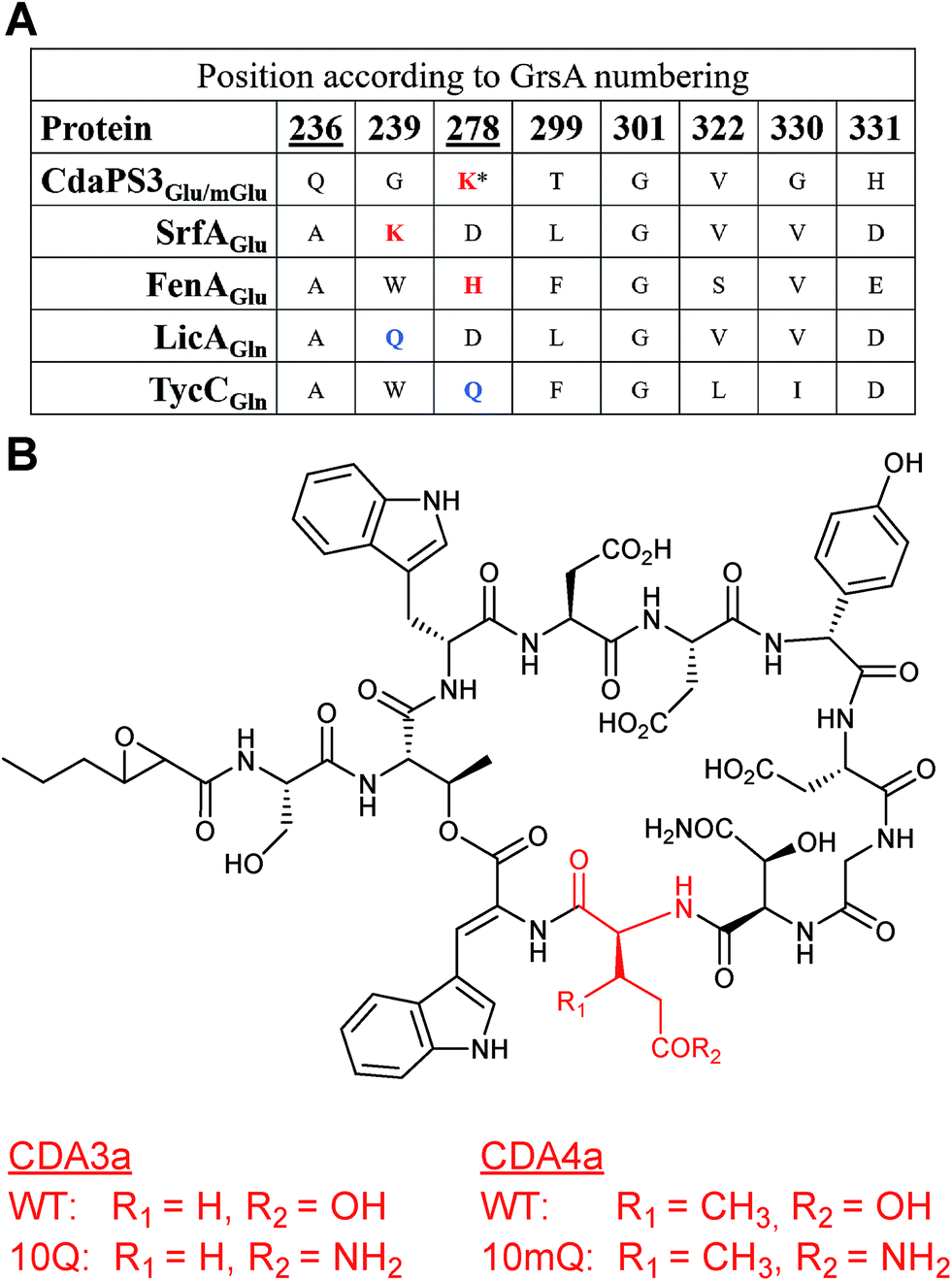 | ||
| Fig. 19 (A) Sequence alignment of specificity-conferring codes for the Glu-specifying A domains of CdaPS3, SrfA and FenA and the Gln-specifying A domains of LicA and TycC, with positions relative to those in GrsA. Residues targeted for mutation in CdaPS3 are indicated with an underscore, red text indicates a basic K or H at positions 239 or 278 in Glu-specifying A domains and blue text indicates an uncharged Q at the same positions in Gln-specifying A domains. * indicates the residue in CdaPS3 mutated from K to Q leading to production of CDA derivatives. (B) Structures of CDA lipopeptides showing the native Glu/mGlu residues and the non-native (10Q and 10mQ) Gln/mGln residues incorporated at position ten, the latter of which were produced by S. coelicolor following K278Q mutation of the CdaPS3 A domain.67 | ||
Since then, two further examples where residues in the A domain binding pocket were mutated to promote the introduction of alternative and non-natural amino acids have been reported. The first of these in 2014 utilised the well-characterised Phe-specifying A domain of GrsA (DAWTIAAICK), in which the eight variable residues that recognise the substrate were chosen to create a library containing single point mutations in the A domain. Each of these mutants were expressed in E. coli and purified for use in a 96 well plate PPi exchange assay utilizing each of the 20 proteinogenic amino acids. Mutation of one residue at position 239 from a Trp to a Ser altered the substrate preference from L-Phe towards L-Tyr. Subsequent modelling suggested this was due to the mutation increasing the volume of the active site cavity. The mutant A domain was then shown to not only possess the ability to adenylate para-substituted phenylalanine derivatives but also the functionalisable ‘clickable’ amino acids p-azido-L-Phe and O-propargyl-L-Tyr (79) with high efficiency.69 When the mutant A domain was expressed, together with the adjacent T–E domains, alongside the first module of GrsB, the ability to form the diketopiperazine (DKP) product, O-propargyl-L-Tyr-L-Pro (81), was demonstrated (Fig. 20). Furthermore, the same mutation was transferred to TycA, and DKP formation and was shown both in vitro as well as in vivo, the latter of which indicates that this mutant may be able to function during nonribosomal peptide biosynthesis to allow labelling of nonribosomal peptides by click chemistry which may, for example, be used to increase bioactivity and broaden structural diversity.
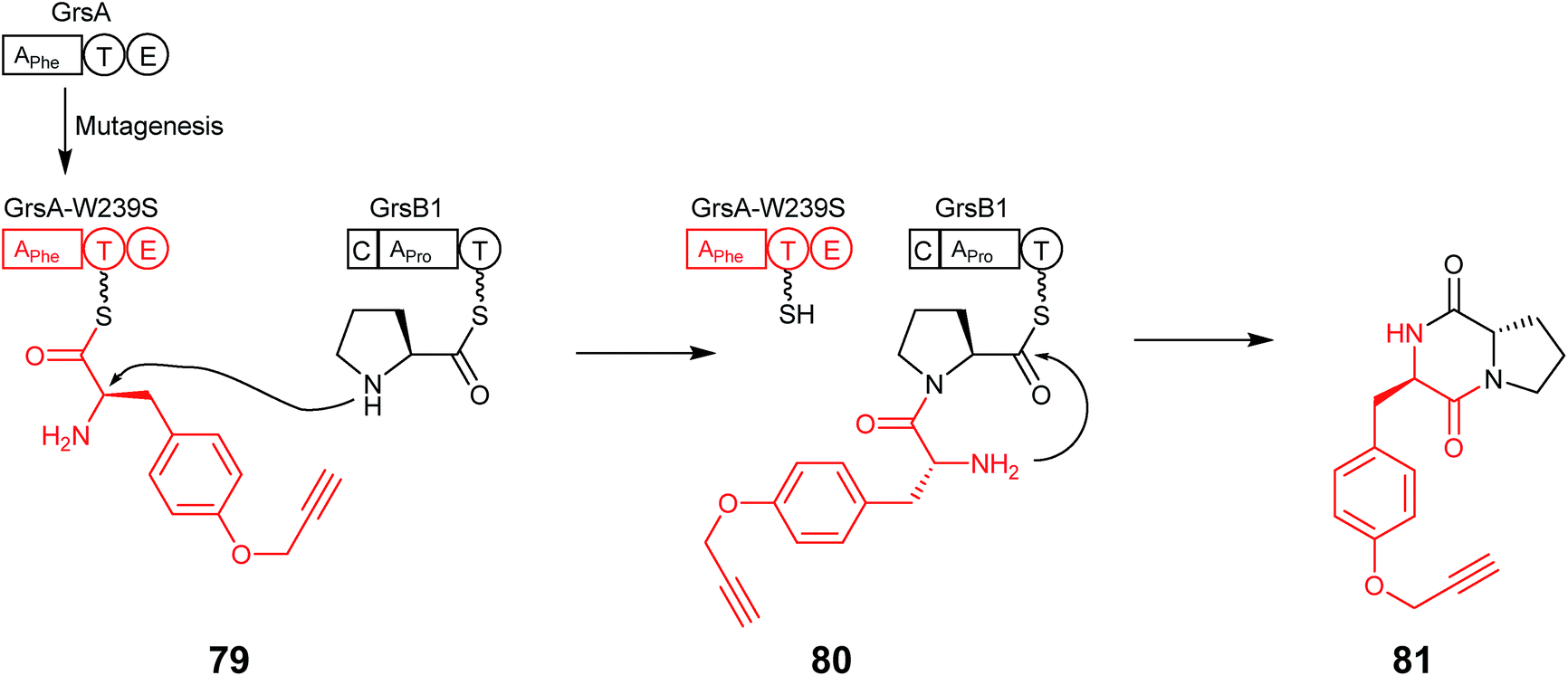 | ||
| Fig. 20 GrsA-W239S mutant with the ability to adenylate the non-natural O-propargyl-L-Tyr (79). Coupling this mutant with GrsB1 allowed the formation of the diketopiperazine O-propargyl-L-Tyr-L-Pro (81).69 | ||
A second example of an A domain being engineered to allow the introduction of azide-containing amino acid derivatives was reported recently.70 In this study, the structural basis for the ability of an A domain to simultaneously recognise two different amino acids, Arg and Tyr, that are incorporated into anabaenopeptin was determined by solving the structure of the A domain in complex with each amino acid. Based on the structural information obtained, it was theorised that the mutation of amino acids at three positions (E204, S243, and A307) could lead to formation of an A domain with a shifted specificity towards Tyr. Following mutations at position 307 only 4 out of 19 mutations were active in a PPi exchange assay. A switch in substrate preference towards Tyr, relative to Arg, seemed to be most favoured when a large aliphatic side chain-containing amino acid was present at position 307 (Val or Leu). Mutations at position 243 produced 15 active mutants out of 19 mutations with large non-polar side chain-containing amino acids again selecting Tyr over Arg. Mutation of the Glu at position 204 in the A domain revealed that this was key for Arg selection, with mutations at this position accepting only tyrosine.70 A new substrate preference for Trp was also observed with the S243E mutant and the double mutant (E204G/S243E) was shown to actually prefer Trp as a substrate. Several of the single mutants also demonstrated the ability to activate Trp as well as the unnatural 4-azido-Phe (Az). Importantly the double mutant (E204G/S243E) was shown to be capable of using Az as a substrate with activity that was similar to the wild-type A domain with Arg.
5.3 Directed evolution of NRPS module specificity
An interesting point that the authors of the previous study noted is that variations in two of the residues identified as important for bispecificity of the A domain in anabaenopeptin production have been found to occur naturally within the Planktothrix genus, and that one of these specifically incorporates Arg.70 Much evidence exists to suggest that the huge number of nonribosomal peptide biosynthetic pathways found in nature evolved via point mutations, genetic duplication, deletion and insertion events (which are especially prominent within the modular NRPS-encoding genes).71 How these NRPS modules maintained, modified or gained the ability to function and produce new compounds is the basis for an attractive approach to engineering NRPS pathways. Through directed evolution, mutations are introduced into an NRPS-encoding gene in the pathway of interest, often targeting the portion of the A domain harbouring the residues of the specificity conferring code. The first example of this directed evolution approach being applied to the improvement of nonribosomal peptide production was shown following transplantation of the first A domain from SyrE (that incorporates Ser1 into syringomycin) in place of the native Ser-specifying A domain of EntF from the enterobactin (82) biosynthetic pathway (Fig. 21). Despite the fact that both adenylation domains incorporate L-serine, a drastic 30 fold loss in activity was observed with the chimeric domain, presumably due to the usual problems of incompatible linking regions that have been addressed above. The authors therefore turned their attention to improving the activity through the application of directed evolution. Mutagenic PCR was used to introduce random mutations into the transplanted SyrE adenylation domain and a small library of mutants was created. As enterobactin is an iron-scavenger the efficiency of each mutant was assessed by growth on low iron media. 28 of the colonies that showed the best growth were taken forward into a subsequent round of screening and the best candidate from those subsequent screens was put through a second round of mutagenesis and screening until mutants were identified that grew as well as the wild-type EntF strain. Three SyrE-EntF mutant adenylation domains selected following both rounds of screening were expressed in E. coli and purified alongside wild-type EntF and the non-mutated SyrE-EntF chimera for use in an in vitro assay of activity. The best mutant following the first round of screening displayed a 2-fold increase in activity over the non-mutated chimera, however the mutants selected following the second round of screening showed larger improvements in activity of around 3-and 8-fold, with the latter being only around 4-fold lower than wild-type EntF.72 Both of these mutated chimeric A domains contained only 4 amino acid substitutions relative to the original chimera and demonstrated the relative ease with which directed evolution can be used to restore function to a chimeric NRPS.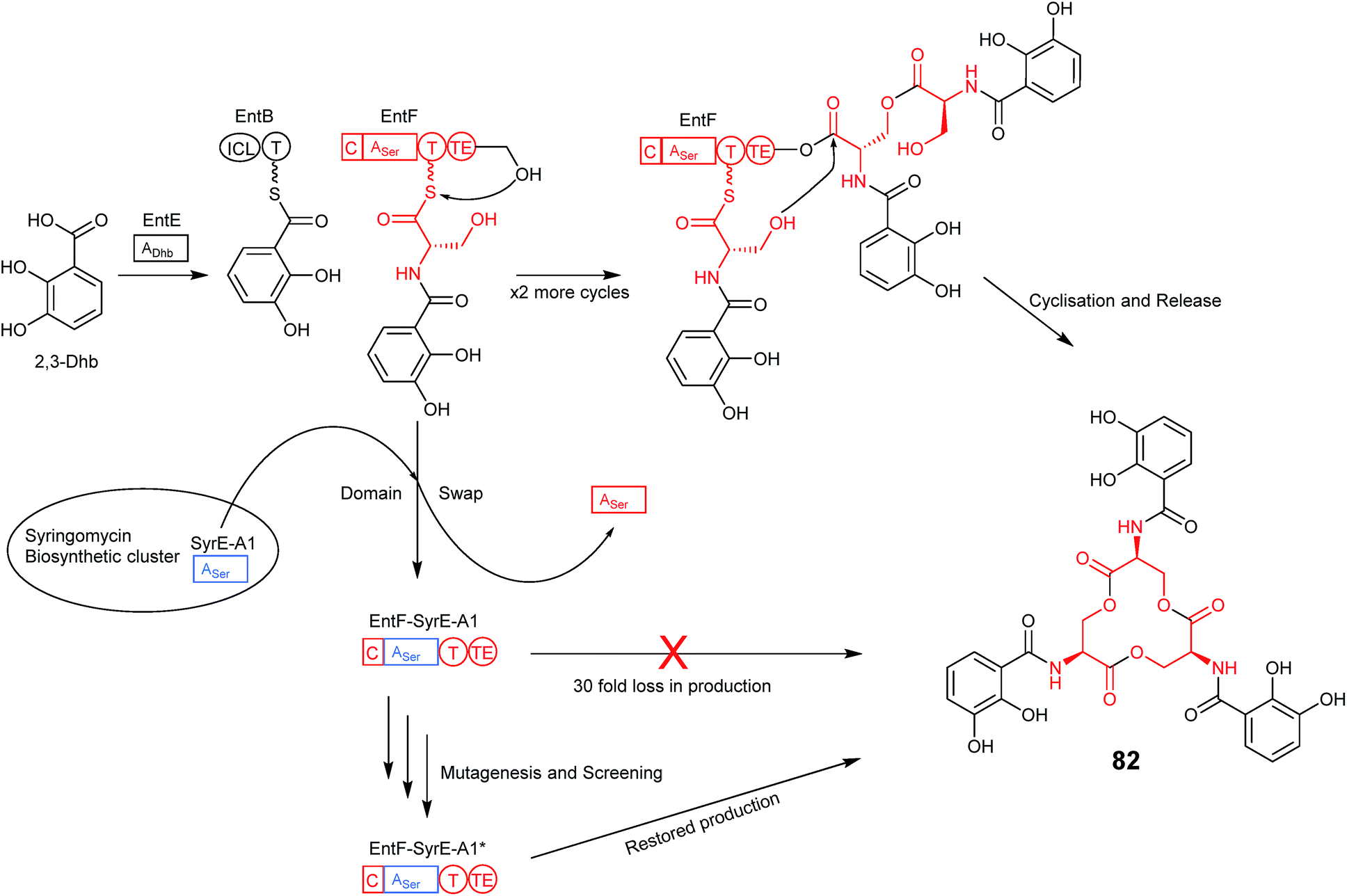 | ||
| Fig. 21 The serine specific A-domain EntF, from enterobactin biosynthesis, was replaced with a similar serine-specific domain, SyrE, from syringomycin biosynthesis. The new domain was 30 fold less active than the native domain. Subsequent rounds of random mutagenesis and directed evolution restored production to almost wild type levels with only 4 amino acid substitutions needed.72 ICL – isochromate lyase – responsible for biosynthesis of 2,3-dihydroxybenzoate. The standalone EntE A-domain is responsible for loading of Dhb onto EntB. | ||
The authors also demonstrated that directed evolution can be applied to an adenylation domain involved in andrimid production, a NRPS/PKS hybrid. The insertion of the promiscuous A domain CytC1 led to a non-functional chimera, as was expected. Through three rounds of mutagenesis production of andrimid was be restored to only 3-fold less than wild-type and the promiscuous nature of CytC1 was used to produce andrimid analogues containing non-native L-2-aminobutyrate or D-2-aminobutyrate following exogenous feeding. In a second experiment with the andrimid NRPS, an A domain was transplanted that was designed to change the specificity from isoleucine to valine, producing a derivative of andrimid known to have more potent antibacterial activity. This substitution produced the expected product at levels that were 7-fold less than wild-type. Following only a single round of mutagenesis one clone restored production of the andrimid derivative to wild-type levels.72 Mutations that increased production were found to be located not only within the active site of the A domain but also in other distil regions that were not predicted, further demonstrating the advantages of using directed evolution when engineering nonribosomal peptides for in vivo production of novel compounds.
In another study on andrimid published in 2011, error prone PCR was used to introduce a combination of mutations at three specific sites in the A domain, identified by multiple sequence alignment, to produce a library of 14330 mutants.73 These were grown in 96 well plates, pooled and assessed for production of new andrimid derivatives by LC-MS. Four clones were found with altered production profiles (following pooled row and column searches), two of which incorporated Ile or Leu instead of Val and the other two producing both Ala and Phe-containing andrimid derivatives. When assessed for bioactivity, two of these andrimid derivatives had lower MICs than andrimid itself against some of the organisms tested.73 These were double, triple or quadruple mutants, with the latter containing an additional spontaneous mutation that showed high activity and improved solubility. This again indicates the potential importance of residues outside the A domain active site.
In a third study, the Phe-specifying A domain from the tyrocidine synthetase was subject to saturation mutagenesis to introduce mutations into each of the eight non-conserved active site residues, producing eight individual libraries consisting of 45 clones at each position. As the wild-type protein is known to have weakly promiscuous activity for L-Thr, enhanced activation of this amino acid was detected using a PPi exchange assay with L-Thr as a substrate in 96 well plates. From this, two mutations (A301C and C331I) were identified as having a positive influence on activation of L-Thr and were therefore combined, activity assessed and entered into a second round of saturation mutagenesis.74 Two further mutations (I330V and W239M) led to enhanced activity in the PPi exchange assay and these two mutations were therefore combined to give a quadruple mutant which was entered into a third round of saturation mutagenesis. Further rounds of evolution contributed no further improvements to the selectivity towards threonine. The mutant containing three mutations (A301C/C331I/I330V) was found to have the highest catalytic efficiency for L-Thr, with a 12-fold increase compared to wild-type. The catalytic efficiency of this mutant adenylation domain was further assessed with nine substrates that ranged in size. Efficiency was found to increase with decreasing substrate size indicating that a steric effect was conferred by the introduced mutations. As three substrates (L-Val, L-2-amino-butyric acid and L-Ala) showed efficiency that were as good as native A domains for those amino acids, the authors demonstrated that the introduced mutations have led to a novel promiscuous A domain.74
In the final and most recent example of directed evolution of NRPS adenylation domains, a different approach was used to identify and select mutants through the combined use of yeast cell surface display and fluorescence activated cell sorting (FACS).75 Firstly a system normally used with antibodies was adapted to display the wild type A domain of DhbE on the yeast cell surface, which was confirmed by detection of two different fluorescent labels fused to the N- and C-terminals of the protein. Having successfully shown this, a method of detecting substrate binding was demonstrated by preparing a chemical probe that mimics acyl-adenylate form of the substrate, acyl-AMS (AMS is adenosine monosulfamate, an isostere of AMP), and has the ability to be labelled with biotin (Fig. 22A). The biotin tag was detected with phycoerythrin (PE)-labelled streptavidin. It was successfully shown that it was possible to detect both the C-terminal myc tag on the wild-type A domain (indicating production of a full length protein) and the probe (indicating binding of the substrate, or a salicylic acid substrate mimic to the A domain) (Fig. 22B). This system was then used to assess a library of DhbE A domain mutants, created by randomization at four positions (His234, Asn235, Ala333 and Val337) that were shown in the crystal structure of DhbE to interact with the 2-OH group of the natural substrate 2,3-dihydroxybenzoic acid (DHB) but are absent from the desired new substrates 3-hydroxybenzoic acid (3-HBA) and 2-aminobenzoic acid (2-ABA). Following five rounds of cell selection, based on detection of probe binding, detection of N- and C-terminal tags and the ability to bind probe at lower concentration, thirty clones were selected for sequencing. Within these clones there were found to be eight distinct mutants with a combination of mutations in the A domain at three positions, with H234 being mutated to Trp in all cases. Four of these eight mutants were assessed in a PPi exchange assay and it was found that only one had higher catalytic activity than wild-type for 3-HBA but upon further assessment this mutant A domain was found to abolish the ability to transfer 3-HBA to the aryl carrier protein (ArCP) of DhbB despite the ability of the wild-type protein to load around 10%. The structure of the EntE, a related A domain, provided a clue as to why this may be; changes at position 234 may hinder the approach of the PPT arm, thus preventing transfer of the substrate. This mutation was changed back to His and upon reassessment of all four mutants they were found to catalyse adenylation of 3-HBA, with the best of these then being shown to load approximately 60% onto the ArCP of DhbB. Kinetic assessment of this mutant in a PPi release assay revealed a change in specificity of around 30-fold. A similar process using a 2-ABA analogue as the probe led to the identification of a mutant that showed a 206-fold change in substrate specificity when compared to the wild-type A domain. The authors noted that whilst binding of the substrate to the A domain may be improved using this method that does not necessarily translate to improved catalytic activity of the enzyme. However, this method is a novel and efficient way to select mutants with improved binding and potentially improved ability for incorporation of non-native amino acids to create novel nonribosomal peptides.75
 | ||
| Fig. 22 (A) Chemical structure of AMS-biotin conjugated SA (salicylic acid) showing SA in red, AMS in blue, the long flexible linker in magenta and biotin in black. (B) Schematic representation of yeast cell surface display of mutant A domains harbouring an N-terminal Aga2p tag that forms disulphide bridges with Aga1p on the cell surface. Mutants that bind an amino acid substrate mimic (acyl-AMS) tagged with biotin are detected following incubation with a streptavidin–phycoerythrin (strep–PE) conjugate, leading to fluorescence-activated cell sorting (FACS) and identification of mutants showing preference for the substrate tested.75 | ||
5.4 Perspective and future of NRPS directed evolution
It is clear that the work presented here only represents a limited number of examples compared to the large diversity of nonribosomal peptides produced in nature but one recurring observation is that when changes to individual or multiple amino acids are made to an adenylation domain it cannot always be accurately predicted how these introduced mutations will influence the performance of the protein and ultimately the end product. However as the availability of new technologies increases so does the speed at which mutants of interest can be identified and with high-throughput techniques, such as those carried out in 96 well plates, comes the potential for further automation, which should allow us to continue to expand our knowledge of NRPS enzyme complexes and allow us to design chimeric systems in a more truly combinatorial fashion.6 Synthetic biology tools and technologies for re-programming NRPS assembly lines
Despite all the studies presented above, progress in engineering NRPS in vivo has not been rapid. This can partly be attributed to the fact that the traditional techniques for engineering NRPS assembly lines in the native host can be laborious, low throughput, and low yielding. However, there have been some significant developments in synthetic biology in the last few years that hold the potential to speed up the process of NRPS engineering, enabling a higher number of new assembly lines to be created and optimised. These tools are not just applicable to NRPS engineering but have been more widely applied to all natural products.6.1 Sequencing and bioinformatic analysis
At the forefront of these new tools and technologies is the rapid development of next-generation sequencing. The generation of a whole genome sequence has never been more affordable and is enabling research groups to obtain the genome sequence of their own favorite organisms which has consequently led to an increase in the total number of genome sequences available in GenBank.76 In conjunction with this genome data, advanced software tools have been developed for genome-wide prediction of possible biosynthetic gene clusters, such as NRPSpredictor,62 cluster finder77 and the widely used antibiotics and secondary metabolites analysis shell (antiSMASH), which is now in its third iteration.78 These tools can be used to detect putative gene clusters in a sequenced genome, identify nearby genes encoding tailoring enzymes and also highlight any similarities to biosynthetic gene clusters with known end products. However it should be noted that, at this time, despite significant advances in automatic software cluster annotation it is often still important to visually inspect sequencing information and manually assign clusters and cluster boundaries that the automatic software may have missed.6.2 Heterologous expression hosts
Genome analysis has revealed the huge potential that is contained within organisms such as Actinobacteria, Burkholderia and fungal species to produce a number of varied nonribosomal peptides. However, despite a wide range of culturing conditions, co-culture approaches, and advances in the engineering of transcriptional machinery, only a tiny fraction of these can be expressed under laboratory conditions.79 Therefore, the heterologous expression of gene clusters of interest in optimised host strains is a practical alternative for identifying compounds of novel biosynthetic gene clusters in combination with sensitive mass spectrometry (MS) detection tools.Recently there has been a drive to establish optimised, engineered Streptomyces host expression platforms, in which the principal endogenous biosynthetic gene clusters have been deleted. These include the engineered model strain S. coelicolor M1152 and the genome-minimized industrial strain S. avermitilis SUKA.80–82 Of course heterologous expression systems have been developed in other organisms including the yeast Hansenula polymorpha,83Bacillus subtilis84,85 and even E. coli where biosynthetic gene clusters for nonribosomal peptides such as echinomycin, valinomycin, and alterochromides have been successfully expressed.86–88
6.3 DNA assembly tools
The rapidly expanding library of gene cluster information and increased knowledge of the mechanisms involved in nonribosomal peptide biosynthesis are opening the doors for synthetic biology and reconstitution of new assembly lines, bringing together elements from many different NRPS systems. Recent advances in DNA assembly technologies are allowing heterologous genes from different pathways to be expressed together in host strains to produce structural analogues or novel compounds. Techniques for the assembly of individual genes, entire biosynthetic pathways and even the whole genome from small fragments (which can be prepared by PCR, subcloning, or chemical synthesis) have been developed and are now widely used.In the case of Gibson assembly, the linearized target vector and the PCR fragments or chemically synthesized DNA parts containing overlapping sequences are mixed together in a single tube with an exonuclease, which chews-back the fragment from the 5′ to 3′ end. Phusion polymerase then fills in the gaps and a ligase seals the remaining single stranded gaps (Fig. 23).90 This can be used to simply insert a single gene into a vector but has also been used to assemble entire gene clusters, such as the 72-kb pristinamycin PII polyketide biosynthetic gene cluster, which was assembled from 14 individual DNA fragments of 4–5 kb in length. This allowed the authors to insert an additional PII biosynthetic gene cluster into the native host strain, which had the consequence of increasing the production of PII by 45%.93
 | ||
| Fig. 23 Schematic representation of various DNA assembly techniques. | ||
These homology-based assembly methods have also been used for the direct capture of entire gene clusters from pure genomic DNA or even from environmental DNA samples. The most common method is to co-transform a linear cloning vector, flanked with homologous arms, with the target genomic DNA into either yeast (TAR) or engineered Escherichia coli (LLHR).92,94 This engineered E. coli strain combines the traditional λ-red recombination system with two functionally similar proteins from the Rac prophase (RecET). Using this strain all ten megasynthase clusters (ranging from 10–52 kb in length), of unknown function, from restriction digested Photorhabdus luminescens genomic DNA were successfully captured.94 Heterologous expression of two of these clusters identified them as producing the nonribosomal peptides luminmide A/B and the NRPS/PKS hybrid luminmycin A respectively.
TAR cloning has been used for the capture of the 67-kb biosynthetic gene cluster responsible for the biosynthesis of the dichlorinated lipopeptide antibiotic taromycin A95 and a 67-kb amicoumacin NRPS/PKS cluster.84 It has also been applied in the successful reassembly of a 90 kb gene cluster from environmental DNA.96 However, the maximum size limitation for direct capture from genomic DNA has yet to be determined. As well as in vivo methods of recombination, a similar approach has been demonstrated with digested genomic DNA coupled with Gibson assembly to build a final cyclic product in an in vitro manner.97
While incredibly useful, assembly methods based on homologous recombination have limitations, especially if there are repeated sequences or stable secondary structure of single stranded DNA at the end of the fragments to be assembled. These repeated sequences are commonly found in NRPS genes. These will compete with the required single-stranded DNA fragment or hinder the assembly process, greatly reducing the efficiency or even introducing unwanted errors into the assembly.
6.4 Refactoring pathways
DNA assembly approaches can also be applied to the refactoring of biosynthetic gene clusters by introducing strong or inducible promoters in front of genes, deleting negative regulators, and reprogramming the biosynthesis pathway by assembly of hybrid assembly lines. For example the silent spectinabilin gene cluster has been refactored using a DNA Assembler-based “plug-and-play” scaffold by removing all native regulatory control elements and replacing these with a series of constitutive and inducible heterologous promoters. Production of the previously silent end product was detected following fermentation.102Using a combination of these techniques can enable combinatorial biosynthesis of natural product clusters. For example a homologous recombination method such as TAR or Gibson could be used to capture an entire gene cluster from isolated genomic DNA or environmental libraries and then a specific module of interest could be flanked with phage integrase sites to enable that module to be exchanged for entire libraries of different modules to enable true combinatorial biosynthesis.
6.5 Improved selection of mutants
Mutagenesis can be used to great effect in generating chimeric NRPS systems. However while current methodologies are often effective, mutagenesis in a protein coding region is time consuming and domain swapping often involves the insertion of selectable markers which can leave residual scar sequences that could interfere with protein expression. To solve this problem, a modified ccdB-based counter-selection technique was developed which performs a seamless point mutation when combined with oligonucleotide-mediated recombination.103 This method has already been successfully applied to introduce point mutations to an A domain in the biosynthesis of luminmide, a complex nonribosomal peptide produced by Photorabdus luminescens.66 In addition several modifications to the standard Streptomyces method of recombination-mediated mutations have been made to improve the efficiency of the double-crossover events required for a successful gene modification. In the standard paradigm used for gene disruptions or insertions in Streptomyces, the targeted sequence on the host genome is switched for a replacement cassette, usually an antibiotic resistance marker, by homologous recombination from a non-replicative vector (which contains a different resistance marker). Successful mutants are screened to ensure they carry the introduced resistance marker but are sensitive to the vector carried marker, indicating that a double-crossover event has occurred.104 The second cross-over step is more difficult than the first and in strains with a low level of homologous recombination this screening can be time consuming. In an attempt to solve this, a system which uses the meganuclease I-SceI from Saccharomyces cerevisiae is available to more accurately select for double cross-over mutants.105 I-SceI is able to cut double stranded DNA at a specific recognition sequence that is not found naturally in actinomycete genomes and limits the viability of single-cross over clones. With a similar aim a blue-white screen based on an indigoidine synthetase gene reporter has also been effectively demonstrated to identify single-crossover mutants as blue and successful double crossovers as white colonies.1066.6 Genome editing
In the last few years techniques for in vivo targeted genome editing, such as TALEN and CRISPR/Cas have become available for use in Streptomyces.107,108 These tools hold great potential for the editing and optimization of NRPS biosynthetic pathways. Cobb et al. developed a modified CRISPR/Cas system for rapid genome editing of Streptomyces, with efficiency ranging from 70 to 100%, including the deletion of the entire 31 kb red cluster from Streptomyces lividans.108 A similar system, termed CRISPRi, based on a catalytically dead variant of Cas9, was also shown to be efficient at reversibly controlling expression of target genes.1097 Conclusions: summary, opinions and perspective
Over the last ten years there has been significant progress in engineering the biosynthesis of new nonribosomal peptide natural products. Precursor directed biosynthesis and mutasynthesis have been successful in broadening the chemical diversity of nonribosomal peptides. Whilst these techniques are largely limited to conservative modifications, they remain important as a rapid method for the generation of natural product analogues. In addition, new approaches are becoming available allowing for the introduction of more significant changes to the structure of nonribosomal peptides. The development of next generation DNA sequencing technologies is probably the single most important development for the engineering of nonribosomal peptides. The discovery of novel and promiscuous tailoring enzymes encoded in new gene clusters is constantly improving the ways in which in vivo modifications can be made to nonribosomal peptides, with new glycosylation, halogenation, and sulfation enzymes being applied outside of their native clusters to good effect.The creation of larger libraries of NRPS encoding genes is empowering an improved understanding of how these complex assembly line enzymes function and advancing us towards a more combinatorial biosynthetic approach. Despite a large number of good examples of NRPS engineering, progress towards combinatorial nonribosomal peptide biosynthesis has been slow. Early attempts to exchange NRPS domains and modules showed mixed results, with some major successes but many more examples where the same approach was not as effective. Gradually the methods of domain and module exchange have become more surgical but a true understanding of how to reprogram nonribosomal peptide synthetases, whilst maintaining activity comparable to the wild type enzymes, seems to be some way off. As illustrated by the work of Cubist on daptomycin, NRPS module exchanges between biosynthetic gene cluster of very close evolutionary origins can lead to functional chimeric nonribosomal synthetases which retain good activity. However, following too closely the evolutionary relationships between NRPS enzymes, and making obvious modular exchanges, runs the risk of re-creating nonribosomal peptide variants that nature has already sampled and discarded due to sub-optimal biological activity. Clearly the bigger goal for the field is to develop strategies that can allow NRPS re-programming to include new functionality, chemistry that nature is yet to sample, within nonribosomal peptide scaffolds. To this end there is hope with the excellent range of new DNA assembly and editing technologies becoming available, particularly CRISPR/Cas9, that promise to enable rapid changes within NRPS modules. Combined with ever decreasing costs of gene synthesis, the new assembly and editing techniques could allow for a far greater number of mutant and chimeric NRPS constructs to be generated and tested, than was possible by conventional genetic techniques. Should these advances be fully exploited, the rules that govern the architecture of NRPS will become more evident and more radically altered nonribosomal peptides may ultimately be produced. The ultimate goal in engineering of nonribosomal peptides is often suggested to be attainable through a so called “plug-and-play” approach, whereby bespoke modules can be assembled together with characterised linker regions, allowing peptide scaffolds to be assembled in order. However, early attempts at “plug-and-play” have not proved to be as simple as some would like to admit; there remains some significant and exciting work to be done relying on traditional enzymology and structural biology, combined with the major technological advancements.
8 Acknowledgements
The authors' laboratory work is supported by BBSRC grants (BB/K002341/1 and BB/L002299/1). Thanks to Dr Sarah Shepherd for careful proof reading.9 Notes and references
- A. Markham, Drugs, 2014, 74, 1823–1828 CrossRef CAS PubMed.
- G. Weber, K. Schorgendorfer, E. Schneider-Scherzer and E. Leitner, Curr. Genet., 1994, 26, 120–125 CrossRef CAS PubMed.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, EMBO J., 1997, 16, 4174–4183 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS PubMed.
- L. Du and B. Shen, Curr. Opin. Drug Discovery Dev., 2001, 4, 215–228 CAS.
- R. Finking and M. A. Marahiel, Annu. Rev. Microbiol., 2004, 58, 453–488 CrossRef CAS PubMed.
- P. J. Belshaw, C. T. Walsh and T. Stachelhaus, Science, 1999, 284, 486–489 CrossRef CAS PubMed.
- U. Linne, S. Doekel and M. A. Marahiel, Biochemistry, 2001, 40, 15824–15834 CrossRef CAS PubMed.
- F. G. Healy, M. Wach, S. B. Krasnoff, D. M. Gibson and R. Loria, Mol. Microbiol., 2000, 38, 794–804 CrossRef CAS PubMed.
- D. P. O'Brien, P. N. Kirkpatrick, S. W. O'Brien, T. Staroske, T. I. Richardson, D. A. Evans, A. Hopkinson, J. B. Spencer and D. H. Williams, Chem. Commun., 2000, 103–104, 10.1039/a907953j.
- L. Li, W. Deng, J. Song, W. Ding, Q. F. Zhao, C. Peng, W. W. Song, G. L. Tang and W. Liu, J. Bacteriol., 2008, 190, 251–263 CrossRef CAS PubMed.
- I. Molnar, T. Schupp, M. Ono, R. Zirkle, M. Milnamow, B. Nowak-Thompson, N. Engel, C. Toupet, A. Stratmann, D. D. Cyr, J. Gorlach, J. M. Mayo, A. Hu, S. Goff, J. Schmid and J. M. Ligon, Chem. Biol., 2000, 7, 97–109 CrossRef CAS PubMed.
- C. T. Walsh, H. Chen, T. A. Keating, B. K. Hubbard, H. C. Losey, L. Luo, C. G. Marshall, D. A. Miller and H. M. Patel, Curr. Opin. Chem. Biol., 2001, 5, 525–534 CrossRef CAS PubMed.
- D. F. Ackerley and I. L. Lamont, Chem. Biol., 2004, 11, 971–980 CrossRef CAS PubMed.
- R. Traber, H. Hofmann and H. Kobel, J. Antibiot., 1989, 42, 591–597 CrossRef CAS PubMed.
- R. Thiericke and J. Rohr, Nat. Prod. Rep., 1993, 10, 265–289 RSC.
- Z. Hojati, C. Milne, B. Harvey, L. Gordon, M. Borg, F. Flett, B. Wilkinson, P. J. Sidebottom, B. A. M. Rudd, M. A. Hayes, C. P. Smith and J. Micklefield, Chem. Biol., 2002, 9, 1175–1187 CrossRef CAS PubMed.
- S. Weist, B. Bister, O. Puk, D. Bischoff, S. Pelzer, G. J. Nicholson, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed., 2002, 41, 3383–3385 CrossRef CAS.
- B. Amir-Heidari, J. Thirlway and J. Micklefield, Org. Biomol. Chem., 2008, 6, 975–978 CAS.
- S. Weist, C. Kittel, D. Bischoff, B. Bister, V. Pfeifer, G. J. Nicholson, W. Wohlleben and R. D. Süssmuth, J. Am. Chem. Soc., 2004, 126, 5942–5943 CrossRef CAS PubMed.
- S. Grüschow, E. J. Rackham, B. Elkins, P. L. Newill, L. M. Hill and R. J. Goss, ChemBioChem, 2009, 10, 355–360 CrossRef PubMed.
- N. K. O'Connor, A. S. Hudson, S. L. Cobb, D. O'Neil, J. Robertson, V. Duncan and C. D. Murphy, Amino Acids, 2014, 46, 2745–2752 CrossRef PubMed.
- W. Zhang, I. Ntai, M. L. Bolla, S. J. Malcolmson, D. Kahne, N. L. Kelleher and C. T. Walsh, J. Am. Chem. Soc., 2011, 133, 5240–5243 CrossRef CAS PubMed.
- A. D. Roy, S. Grüschow, N. Cairns and R. J. M. Goss, J. Am. Chem. Soc., 2010, 132, 12243–12245 CrossRef CAS PubMed.
- R. H. Baltz, V. Miao and S. K. Wrigley, Nat. Prod. Rep., 2005, 22, 717–741 RSC.
- A. Malina and Y. Shai, Biochem. J., 2005, 390, 695–702 CrossRef CAS PubMed.
- V. M. D'Costa, T. A. Mukhtar, T. Patel, K. Koteva, N. Waglechner, D. W. Hughes, G. D. Wright and G. de Pascale, Antimicrob. Agents Chemother., 2012, 56, 757–764 CrossRef PubMed.
- R. A. Lewis, L. Nunns, J. Thirlway, K. Carroll, C. P. Smith and J. Micklefield, Chem. Commun., 2011, 47, 1860–1862 RSC.
- A. Powell, M. Borg, B. Amir-Heidari, J. M. Neary, J. Thirlway, B. Wilkinson, C. P. Smith and J. Micklefield, J. Am. Chem. Soc., 2007, 129, 15182–15191 CrossRef CAS PubMed.
- E. Higashide, K. Hatano, M. Shibata and K. Nakazawa, J. Antibiot., 1968, 21, 126–137 CrossRef CAS PubMed.
- B. Cavalleri, H. Pagani, G. Volpe, E. Selva and F. Parenti, J. Antibiot., 1984, 37, 309–317 CrossRef CAS PubMed.
- X. Yin and T. M. Zabriskie, Microbiology, 2006, 152, 2969–2983 CrossRef PubMed.
- X. Yin, Y. Chen, L. Zhang, Y. Wang and T. M. Zabriskie, J. Nat. Prod., 2010, 73, 583–589 CrossRef CAS PubMed.
- P. Cudic, D. C. Behenna, J. K. Kranz, R. G. Kruger, A. J. Wand, Y. I. Veklich, J. W. Weisel and D. G. McCafferty, Chem. Biol., 2002, 9, 897–906 CrossRef CAS PubMed.
- M.-C. Wu, M. Q. Styles, B. J. C. Law, A.-W. Struck, L. Nunns and J. Micklefield, Microbiology, 2015, 161, 1338–1347 CrossRef PubMed.
- J. S. Chen, Y. X. Wang, L. Shao, H. X. Pan, J. A. Li, H. M. Lin, X. J. Dong and D. J. Chen, Biotechnol. Lett., 2013, 35, 1501–1508 CrossRef CAS PubMed.
- S. Di Palo, R. Gandolfi, S. Jovetic, F. Marinelli, D. Romano and F. Molinari, Enzyme Microb. Technol., 2007, 41, 806–811 CrossRef CAS.
- J. C. J. Barna, D. H. Williams, D. J. M. Stone, T. W. C. Leung and D. M. Doddrell, J. Am. Chem. Soc., 1984, 106, 4895–4902 CrossRef CAS.
- B. P. Goldstein, E. Selva, L. Gastaldo, M. Berti, R. Pallanza, F. Ripamonti, P. Ferrari, M. Denaro, V. Arioli and G. Cassani, Antimicrob. Agents Chemother., 1987, 31, 1961–1966 CrossRef CAS PubMed.
- S.-Y. Lyu, Y.-C. Liu, C.-Y. Chang, C.-J. Huang, Y.-H. Chiu, C.-M. Huang, N.-S. Hsu, K.-H. Lin, C.-J. Wu, M.-D. Tsai and T.-L. Li, J. Am. Chem. Soc., 2014, 136, 10989–10995 CrossRef CAS PubMed.
- J. J. Banik and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17273–17277 CrossRef CAS PubMed.
- L. D. Boeck and F. P. Mertz, J. Antibiot., 1986, 39, 1533–1540 CrossRef CAS PubMed.
- L. Kalan, J. Perry, K. Koteva, M. Thaker and G. Wright, J. Bacteriol., 2013, 195, 167–171 CrossRef CAS PubMed.
- N. J. Skelton, D. H. Williams, R. A. Monday and J. C. Ruddock, J. Org. Chem., 1990, 55, 3718–3723 CrossRef CAS.
- G. Yim, L. Kalan, K. Koteva, M. N. Thaker, N. Waglechner, I. Tang and G. D. Wright, ChemBioChem, 2014, 15, 2613–2623 CrossRef CAS PubMed.
- R. H. Baltz, P. Brian, V. Miao and S. K. Wrigley, J. Ind. Microbiol. Biotechnol., 2006, 33, 66–74 CrossRef CAS PubMed.
- V. Miao, M. F. Coeffet-Le Gal, K. Nguyen, P. Brian, J. Penn, A. Whiting, J. Steele, D. Kau, S. Martin, R. Ford, T. Gibson, M. Bouchard, S. K. Wrigley and R. H. Baltz, Chem. Biol., 2006, 13, 269–276 CrossRef CAS PubMed.
- M. F. Coëffet-Le Gal, L. Thurston, P. Rich, V. Miao and R. H. Baltz, Microbiology, 2006, 152, 2993–3001 CrossRef PubMed.
- K. T. Nguyen, D. Ritz, J. Q. Gu, D. Alexander, M. Chu, V. Miao, P. Brian and R. H. Baltz, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17462–17467 CrossRef CAS PubMed.
- S. Doekel, M.-F. Coëffet-Le Gal, J.-Q. Gu, M. Chu, R. H. Baltz and P. Brian, Microbiology, 2008, 154, 2872–2880 CrossRef CAS PubMed.
- M. J. Calcott, J. G. Owen, I. L. Lamont and D. F. Ackerley, Appl. Environ. Microbiol., 2014, 80, 5723–5731 CrossRef PubMed.
- H. D. Mootz, N. Kessler, U. Linne, K. Eppelmann, D. Schwarzer and M. A. Marahiel, J. Am. Chem. Soc., 2002, 124, 10980–10981 CrossRef CAS PubMed.
- D. Butz, T. Schmiederer, B. Hadatsch, W. Wohlleben, T. Weber and R. D. Süssmuth, ChemBioChem, 2008, 9, 1195–1200 CrossRef CAS PubMed.
- D. Yu, F. Xu, D. Gage and J. Zhan, Chem. Commun., 2013, 49, 6176–6178 RSC.
- R. Beer, K. Herbst, N. Ignatiadis, I. Kats, L. Adlung, H. Meyer, D. Niopek, T. Christiansen, F. Georgi, N. Kurzawa, J. Meichsner, S. Rabe, A. Riedel, J. Sachs, J. Schessner, F. Schmidt, P. Walch, K. Niopek, T. Heinemann, R. Eils and B. Di Ventura, Mol. BioSyst., 2014, 10, 1709–1718 RSC.
- A. S. Reger, J. M. Carney and A. M. Gulick, Biochemistry, 2007, 46, 6536–6546 CrossRef CAS PubMed.
- R. Wu, A. S. Reger, X. Lu, A. M. Gulick and D. Dunaway-Mariano, Biochemistry, 2009, 48, 4115–4125 CrossRef CAS PubMed.
- B. R. Miller, J. A. Sundlov, E. J. Drake, T. A. Makin and A. M. Gulick, Proteins, 2014, 82, 2691–2702 CrossRef CAS PubMed.
- M. Crüsemann, C. Kohlhaas and J. Piel, Chem. Sci., 2013, 4, 1041–1045 RSC.
- H. Kries, D. L. Niquille and D. Hilvert, Chem. Biol., 2015, 22, 640–648 CrossRef CAS PubMed.
- M. Rottig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed.
- K. Kurusu, K. Ohba, T. Arai and K. Fukushima, J. Antibiot., 1987, 40, 1506–1514 CrossRef CAS PubMed.
- Y. Kajimura and M. Kaneda, J. Antibiot., 1996, 49, 129–135 CrossRef CAS PubMed.
- J. Han, E. Kim, J. Lee, Y. Kim, E. Bang and B. Kim, Biotechnol. Lett., 2012, 34, 1327–1334 CrossRef CAS PubMed.
- X. Bian, A. Plaza, F. Yan, Y. Zhang and R. Müller, Biotechnol. Bioeng., 2015 Search PubMed.
- J. Thirlway, R. Lewis, L. Nunns, M. Al Nakeeb, M. Styles, A. W. Struck, C. P. Smith and J. Micklefield, Angew. Chem., Int. Ed., 2012, 51, 7181–7184 CrossRef CAS PubMed.
- C. Milne, A. Powell, J. Jim, M. Al Nakeeb, C. P. Smith and J. Micklefield, J. Am. Chem. Soc., 2006, 128, 11250–11259 CrossRef CAS PubMed.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 CrossRef CAS PubMed.
- H. Kaljunen, S. H. Schiefelbein, D. Stummer, S. Kozak, R. Meijers, G. Christiansen and A. Rentmeister, Angew. Chem., Int. Ed., 2015 Search PubMed.
- D. E. Cane, C. T. Walsh and C. Khosla, Science, 1998, 282, 63–68 CrossRef CAS PubMed.
- M. A. Fischbach, J. R. Lai, E. D. Roche, C. T. Walsh and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11951–11956 CrossRef CAS PubMed.
- B. S. Evans, Y. Chen, W. W. Metcalf, H. Zhao and N. L. Kelleher, Chem. Biol., 2011, 18, 601–607 CrossRef CAS PubMed.
- B. Villiers and F. Hollfelder, Chem. Biol., 2011, 18, 1290–1299 CrossRef CAS PubMed.
- K. Zhang, K. M. Nelson, K. Bhuripanyo, K. D. Grimes, B. Zhao, C. C. Aldrich and J. Yin, Chem. Biol., 2013, 20, 92–101 CrossRef CAS PubMed.
- D. A. Benson, K. Clark, I. Karsch-Mizrachi, D. J. Lipman, J. Ostell and E. W. Sayers, Nucleic Acids Res., 2014, 42, D32–D37 CrossRef CAS PubMed.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Muller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, 239–243 CrossRef PubMed.
- T. Weber, P. Charusanti, E. M. Musiol-Kroll, X. Jiang, Y. Tong, H. U. Kim and S. Y. Lee, Trends Biotechnol., 2015, 33, 15–26 CrossRef CAS PubMed.
- J. P. Gomez-Escribano and M. J. Bibb, Microb. Biotechnol., 2011, 4, 207–215 CrossRef CAS PubMed.
- M. Komatsu, T. Uchiyama, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2646–2651 CrossRef CAS PubMed.
- M. Komatsu, K. Komatsu, H. Koiwai, Y. Yamada, I. Kozone, M. Izumikawa, J. Hashimoto, M. Takagi, S. Omura, K. Shin-ya, D. E. Cane and H. Ikeda, ACS Synth. Biol., 2013, 2, 384–396 CrossRef CAS PubMed.
- L. Gidijala, J. A. Kiel, R. D. Douma, R. M. Seifar, W. M. van Gulik, R. A. Bovenberg, M. Veenhuis and I. J. van der Klei, PLoS One, 2009, 4, e8317 Search PubMed.
- Y. Li, Z. Li, K. Yamanaka, Y. Xu, W. Zhang, H. Vlamakis, R. Kolter, B. S. Moore and P.-Y. Qian, Sci. Rep., 2015, 5, 9383 CrossRef CAS PubMed.
- S. Zobel, J. Kumpfmüller, R. Süssmuth and T. Schweder, Appl. Microbiol. Biotechnol., 2015, 99, 681–691 CrossRef CAS PubMed.
- K. Watanabe, H. Oguri and H. Oikawa, Curr. Opin. Chem. Biol., 2009, 13, 189–196 CrossRef CAS PubMed.
- J. Jaitzig, J. Li, R. D. Süssmuth and P. Neubauer, ACS Synth. Biol., 2014, 3, 432–438 CrossRef CAS PubMed.
- A. C. Ross, L. E. S. Gulland, P. C. Dorrestein and B. S. Moore, ACS Synth. Biol., 2015, 4, 414–420 CrossRef CAS PubMed.
- M. Z. Li and S. J. Elledge, Nat. Methods, 2007, 4, 251–256 CrossRef CAS PubMed.
- D. G. Gibson, L. Young, R. Y. Chuang, J. C. Venter, C. A. Hutchison 3rd and H. O. Smith, Nat. Methods, 2009, 6, 343–345 CrossRef CAS PubMed.
- Z. Shao, H. Zhao and H. Zhao, Nucleic Acids Res., 2009, 37, e16 CrossRef PubMed.
- N. Kouprina and V. Larionov, Nat. Protoc., 2008, 3, 371–377 CrossRef CAS PubMed.
- L. Li, Y. Zhao, L. Ruan, S. Yang, M. Ge, W. Jiang and Y. Lu, Metab. Eng., 2015, 29, 12–25 CrossRef CAS PubMed.
- J. Fu, X. Bian, S. Hu, H. Wang, F. Huang, P. M. Seibert, A. Plaza, L. Xia, R. Müller, A. F. Stewart and Y. Zhang, Nat. Biotechnol., 2012, 30, 440–446 CrossRef CAS PubMed.
- K. Yamanaka, K. A. Reynolds, R. D. Kersten, K. S. Ryan, D. J. Gonzalez, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1957–1962 CrossRef CAS PubMed.
- J. H. Kim, Z. Feng, J. D. Bauer, D. Kallifidas, P. Y. Calle and S. F. Brady, Biopolymers, 2010, 93, 833–844 CrossRef CAS PubMed.
- Y. Zhou, A. C. Murphy, M. Samborskyy, P. Prediger, L. C. Dias and P. F. Leadlay, Chem. Biol., 2015, 22, 745–754 CrossRef CAS PubMed.
- C. Engler, R. Kandzia and S. Marillonnet, PLoS One, 2008, 3, e3647 Search PubMed.
- S. de Kok, L. H. Stanton, T. Slaby, M. Durot, V. F. Holmes, K. G. Patel, D. Platt, E. B. Shapland, Z. Serber, J. Dean, J. D. Newman and S. S. Chandran, ACS Synth. Biol., 2014, 3, 97–106 CrossRef CAS PubMed.
- L. Zhang, G. Zhao and X. Ding, Sci. Rep., 2011, 1, 141 Search PubMed.
- S. D. Colloms, C. A. Merrick, F. J. Olorunniji, W. M. Stark, M. C. Smith, A. Osbourn, J. D. Keasling and S. J. Rosser, Nucleic Acids Res., 2014, 42, e23 CrossRef CAS PubMed.
- Z. Shao, G. Rao, C. Li, Z. Abil, Y. Luo and H. Zhao, ACS Synth. Biol., 2013, 2, 662–669 CrossRef CAS PubMed.
- H. Wang, X. Bian, L. Xia, X. Ding, R. Muller, Y. Zhang, J. Fu and A. F. Stewart, Nucleic Acids Res., 2014, 42, e37 CrossRef CAS PubMed.
- B. Gust, G. Chandra, D. Jakimowicz, T. Yuqing, C. J. Bruton and K. F. Chater, Adv. Appl. Microbiol., 2004, 54, 107–128 CAS.
- L. T. Fernández-Martínez and M. J. Bibb, Sci. Rep., 2014, 4, 7100 CrossRef PubMed.
- P. Li, J. Li, Z. Guo, W. Tang, J. Han, X. Meng, T. Hao, Y. Zhu, L. Zhang and Y. Chen, Appl. Microbiol. Biotechnol., 2015, 99, 1923–1933 CrossRef CAS PubMed.
- J. K. Joung and J. D. Sander, Nat. Rev. Mol. Cell Biol., 2013, 14, 49–55 CrossRef CAS PubMed.
- R. E. Cobb, Y. Wang and H. Zhao, ACS Synth. Biol., 2014, 4, 723–728 CrossRef PubMed.
- Y. Tong, P. Charusanti, L. Zhang, T. Weber and S. Y. Lee, ACS Synth. Biol., 2015, 4, 1020–1029 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |