
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipid-coated polymeric nanoparticles for cancer drug delivery
Sangeetha
Krishnamurthy
a,
Rajendran
Vaiyapuri
a,
Liangfang
Zhang
b and
Juliana M.
Chan
*ac
aDivision of Bioengineering, School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore. E-mail: julianachan@ntu.edu.sg
bDepartment of NanoEngineering, University of California-San Diego, La Jolla, San Diego, USA
cLee Kong Chian School of Medicine, Singapore
First published on 11th February 2015
Abstract
Polymeric nanoparticles and liposomes have been the platform of choice for nanoparticle-based cancer drug delivery applications over the past decade, but extensive research has revealed their limitations as drug delivery carriers. A hybrid class of nanoparticles, aimed at combining the advantages of both polymeric nanoparticles and liposomes, has received attention in recent years. These core/shell type nanoparticles, frequently referred to as lipid–polymer hybrid nanoparticles (LPNs), possess several characteristics that make them highly suitable for drug delivery. This review introduces the formulation methods used to synthesize LPNs and discusses the strategies used to treat cancer, such as by targeting the tumor microenvironment or vasculature. Finally, it discusses the challenges that must be overcome to realize the full potential of LPNs in the clinic.
 Sangeetha Krishnamurthy | Dr Sangeetha Krishnamurthy received her Master of Science degree in Molecular Sciences and Nanotechnology from Louisiana Tech University in 2011 and went on to receive a Ph.D. in Integrative Sciences and Engineering from National University of Singapore in 2015 under the guidance of Dr Yi Yan Yang from the Institute of Bioengineering and Nanotechnology, Singapore. Currently she is a postdoctoral fellow in Assistant Professor Dr Juliana Chan's group at Nanyang Technological University. Her research interests focus on using biodegradable and biocompatible liposomal and soft polymeric materials for the delivery of therapeutics and antimicrobial applications. |
 Liangfang Zhang | Dr. Liangfang Zhang is a Professor in the Department of NanoEngineering and Moores Cancer Center at the University of California, San Diego. He received his Ph.D. in Chemical Engineering at the University of Illinois at Urbana-Champaign. His research interests focus on the design, synthesis, characterization and evaluation of nanostructured biomaterials for drug delivery to improve or enable treatments of human diseases, with particular interest in cancers and bacterial infections. |
 Juliana Chan | Assistant Professor Juliana Chan is a Nanyang Assistant Professor at Nanyang Technological University and the Lee Kong Chian School of Medicine. She received a Ph.D. in Biology from the Massachusetts Institute of Technology (MIT) under the supervision of Professor Robert Langer. Juliana Chan is a recipient of the 2011 L'Oréal-UNESCO for Women in Science National Fellowships (Singapore) and one of the 10 Innovators Under 35 by MIT Technology Review (Southeast Asia, Australia and New Zealand) in 2014. Her research interests include microfluidics and nanoparticle-based drug delivery. |
1. Introduction
Nanomedicine is a rapidly growing field that uses nanotechnology to solve clinical problems. The National Institutes of Health (NIH) defines nanomedicine as a molecular scale intervention for the prevention, diagnosis and treatment of disease. Currently, there is much interest in the application of nanomedicine for the diagnosis and treatment of cancer, which is linked to high mortality and morbidity rates. According to the World Health Organization, cancer is the second major cause of death from non-communicable diseases worldwide (8.2 million in 2012), next only to cardiovascular diseases.1Indeed, nanomedicine has increasingly been shown to offer various advantages over traditional drug delivery methods. Various properties make them very attractive for the treatment of cancer: these include their small size, high surface area to volume ratio, ability to load multiple agents, active targeting by conjugating targeting ligands on their surface, passive targeting through the enhanced permeability and retention (EPR) effect, and an improved circulation half-life.2
Scientists have developed nanoparticles that can be grouped into several broad classes, each with its own advantages and disadvantages. These include polymeric nanoparticles, liposomes, magnetic nanoparticles, micelles, gold nanoparticles, quantum dots, dendrimers and carbon nanotubes.3 However, most of these nanoparticles are still at the research stage and only a handful of them have been approved for clinical use. The majority of clinically approved formulations are liposomes and these include Doxil and Myocet.3 Clinically approved polymeric nanoparticles include Adagen, Genexol PM, Eligard and Copaxone.3
Although preferred for their excellent biocompatibility, liposomes are associated with challenges such as drug leakage and instability during storage, leading to a short shelf-life.4 Polymeric nanoparticles show excellent loading and stability, but on the other hand may involve complex synthetic procedures and materials, and thus require rigorous biocompatibility testing before they can be brought to the clinic.5 Although some of the main hurdles in designing nanoparticle-based drug delivery carriers have been overcome, the requirements of an ideal cancer drug delivery system become increasingly complicated when the carrier's interactions with the body and the complexity of the disease are also considered.
To broadly fulfill the requirements of high biocompatibility and drug loading capacity, it becomes desirable to combine the advantages of the two dominant classes of drug nanocarrier systems – liposomes and polymeric nanoparticles. Researchers have developed hybrids of these two classes of nanoparticles, broadly referred to as lipid–polymer hybrid nanoparticles (LPNs), and have used them for various therapeutic and diagnostic applications. A typical LPN has a polymeric core, which is used to encapsulate the cargo to be delivered; and a lipid shell, which provides biocompatibility. The lipid shell may either be a monolayer or a bilayer. In some cases, an additional layer of poly(ethylene glycol) (PEG) is further coated onto the lipid surface to give it stealth properties in the bloodstream.6 The work by Miguel and co-workers from 2000 is one of the earliest reported studies on bilayer lipid-coated polymeric nanoparticle synthesis. The bilayer LPNs were synthesized from epichlorohydrin cross-linked polysaccharides modified with quaternary ammonium functions and coated with a layer of lipid and cholesterol.7 Later in 2008, Zhang et al. first described a simple self-assembly method to synthesize the more widely used monolayer lipid-coated polymeric nanoparticles, which showcased monolayer LPNs as a robust drug delivery platform.8
Since then, there has been a large increase in the diversity of biomaterials used for LPN synthesis and applications reported in the literature. This review discusses the various synthetic procedures used to prepare LPNs, the advantages that make LPNs excellent delivery agents for cancer therapy, as well as the applications they have been used in.
2. Preparation of LPNs
In most of the studies described here, the core of the LPN is synthesized from clinically-approved biomaterial poly(lactide-co-glycolide) (PLGA), owing to its biocompatibility and biodegradability.9 As for the lipid shell, various lipids including phosphatidyl choline (PC),10 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE),11,12 cholesterol,13 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC),14 myristic acid,15 stearic acid,16 and 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC)17 have been used.Earlier approaches to LPN synthesis used a two-step procedure, which requires that the lipid shell and the polymeric core be prepared first, followed by a second step to fuse the layers together. In recent years, however, a single-step approach is increasingly preferred, giving researchers greater ease and convenience when preparing LPNs.
2.1. Two-step approach
In the two-step approach, the lipid shell and the polymeric core are prepared individually, before being fused together by direct hydration,18 sonication19 or extrusion,20 to form a bilayer LPN (Fig. 1). Electrostatic interactions between the anionic polymeric core and the cationic lipid vesicle drive the fusion process.21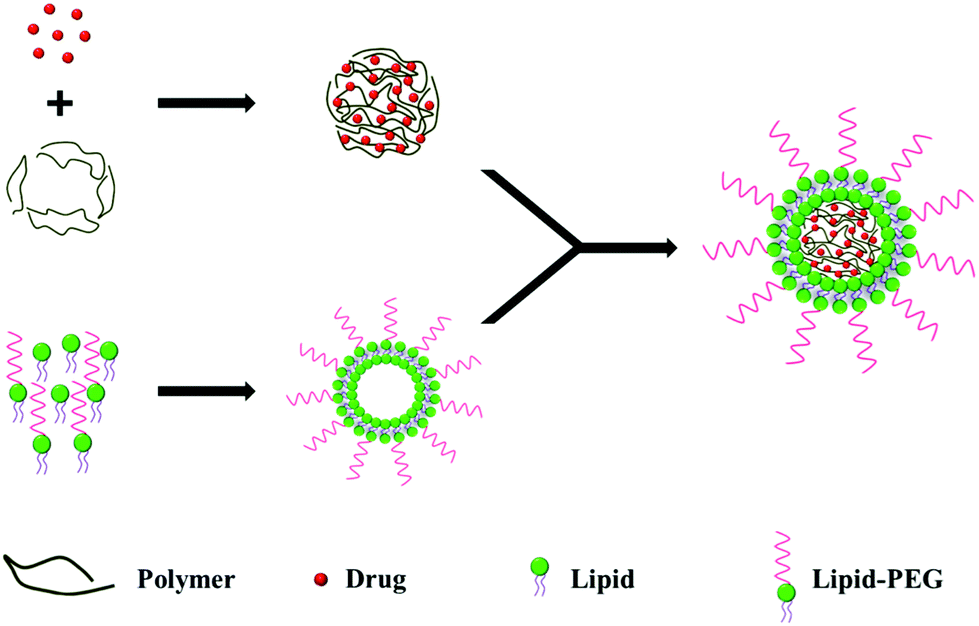 | ||
| Fig. 1 Schematic representation of bilayer LPN preparation using a two-step approach. | ||
Various mixing techniques have been used to fuse the polymeric cores with lipid vesicles. In one example, the solution was heated above the lipid phase transition temperature to disperse the lipid molecules, allowing for the fusion of the polymeric cores with preformed lipid vesicles.21 In another example, the lipids were dried into a film and hydrated in the presence of polymeric nanoparticles.18
Using the two-step approach, Troutier et al. synthesized 500 nm multilamellar lipoparticles by hydration using DPPC, DPTAP and polystyrene. Next, the nanoparticles were sonicated, resulting in multilamellar lipoparticles that were ∼250 nm in diameter. The nanoparticles were further subjected to extrusion to give unilamellar LPNs that were 100 nm in diameter.20 In general, large multilamellar vesicles are formed from the hydration of dry lipid films, which require sonication or extrusion to convert these large multilamellar vesicles into small unilamellar vesicles that are smaller in size and with a lower polydispersity index.
In another study, Sengupta et al. developed a combination therapy system for the dual loading of doxorubicin and combretastatin.22 The shells were formed from phosphatidylcholine, cholesterol, DSPE-PEG and combretastatin, while doxorubicin-PLGA conjugates were loaded into the cores using an emulsion/solvent evaporation method. Finally, the lipid vesicles and polymeric core particles were fused together by extrusion. The nanocells, as they were called, were around 180–200 nm in diameter. They exhibited two different drug release profiles: combretastatin was released quickly from the shell over 12 h, while the doxorubicin-PLGA conjugates were slowly released over 15 days as the polymer degraded and the free drug molecules were released.
In another study, Zhao et al. reported the preparation of folic acid-conjugated LPNs loaded with paclitaxel.18 Here, a thin film of lipids was formed by solvent evaporation and hydrated in a buffer containing paclitaxel-loaded PLGA nanoparticles. The LPNs were approximately 190 nm in diameter.
2.2. Single-step approach
In the single-step approach, the lipid, polymer and drug are mixed together and the monolayer LPNs are prepared by self-assembly (Fig. 2).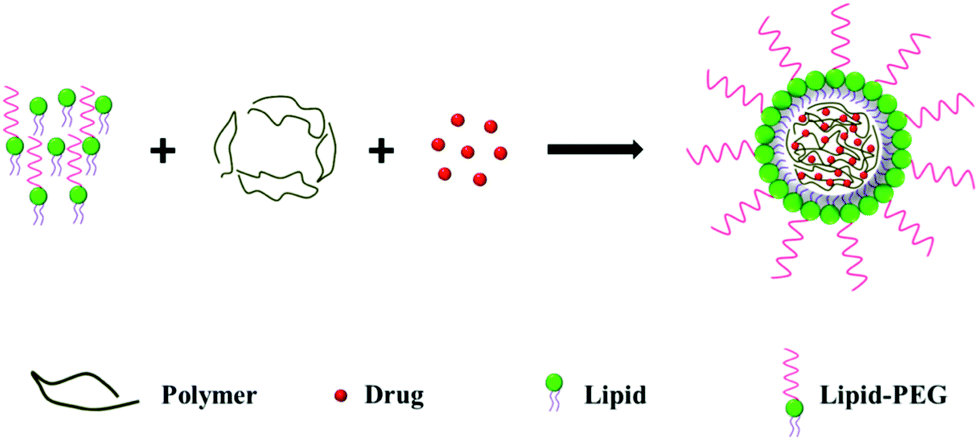 | ||
| Fig. 2 Schematic representation of monolayer LPN preparation using a single-step approach. | ||
One example of the emulsion method was carried out by Palange et al., who synthesized curcumin encapsulated LPNs for breast cancer therapy.23 They prepared an organic solution containing curcumin, PLGA and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), and an aqueous solution containing DSPE-PEG in 4% ethanol solution. The oil phase was added dropwise to the aqueous phase under ultrasonication, producing LPNs that were about 170 nm in diameter.
Using a one-step nanoprecipitation method, Zhang et al. synthesized docetaxel loaded LPNs.8 Here, a 4% ethanol aqueous solution containing lecithin and DSPE-PEG was heated to 65 °C before an organic solution containing PLGA and docetaxel was added dropwise to the aqueous solution under vigorous stirring. The mixture was vortexed vigorously for 3 min, and the organic solvent was removed by gentle stirring for 2 h at room temperature and centrifugal filtration. The resulting particles were about 90 nm in diameter. Similarly, Yang et al. prepared LPNs for the systemic delivery of siRNA using PEG-PLA/PLA and cationic lipid BHEM-Chol.24 In another study, Huang et al. prepared LPNs for dual drug delivery by loading paclitaxel into the hydrophobic core and doxorubicin into the hydrophilic shell.25
3. LPNs for cancer therapy
LPNs have several advantages over other nanoformulations such as liposomes and polymeric nanoparticles, making them good candidates for cancer therapy. Some of their disadvantages are also discussed in section 4.One of the main advantages of using LPNs for cancer drug delivery is the possibility of incorporating drugs with different physicochemical characteristics due to the presence of distinct lipid and polymer layers.26 The ratio of different drugs can be precisely controlled, allowing researchers to co-deliver drugs to cancer cells for a synergistic effect and to overcome multi-drug resistance.27 Hydrophobic drugs can also be co-delivered with novel therapeutic agents such as proteins, peptides and nucleic acids: hydrophobic small molecule drugs can be loaded into the nanoparticle core, while hydrophilic biomolecules can be loaded into the lipid layer or conjugated onto the surface.
LPNs have been shown to provide higher drug loading capacities for hydrophobic drugs compared to polymeric nanoparticles, greater stability without drug leakage, and excellent controlled drug release profiles.8 Their sustained release profile is due to the slow degradation rate of polymers in the core and also the diffusional barrier of the lipid shell. Stimuli-responsive LPNs can be designed to deliver drugs specifically to tumors using pH-sensitive and redox-sensitive LPN systems.28
The lipid shell can be engineered as a monolayer or bilayer, depending upon the release characteristics required and also based on the cargo to be loaded in the lipid shell. In addition, the lipid shell can be functionalized with targeting moieties. By modifying LPNs with various targeting ligands, they can be actively targeted to tumor sites in addition to an EPR effect.11
The synthesis of LPNs, as discussed earlier in section 2, is simple and straightforward, with good control over particle size and ease of surface modification. The PEG layer on the surface of LPNs increases its hydrophilicity, enhancing their circulation half-life and reducing clearance by the reticulo-endothelial system.6
In recent years, several reports using LPNs to deliver therapeutic agents such as cytotoxic drugs, nucleic acids and proteins to tumors have been published. These studies are classified here into seven sections, each representing a possible therapeutic option for the treatment of cancer. They are also summarized in Table 1 for their key characteristics such as delivery of a single agent or combination agents.
| Polymer components | Lipid components | Therapeutic(s) | Targeting | Cancer model | Ref |
|---|---|---|---|---|---|
| Single agent delivery | |||||
| PLGA-PEG-PLGA | Cholesterol, lecithin | Salidroside | N/A | Breast cancer and pancreatic cancer (4T1 and PANC-1 cells) | 13 |
| Hyaluronic acid ceramide (HACE) | Egg PC, DSPE-PEG | Ginsenoside Rg3 | N/A | Lung cancer (A549 cells) | 29 |
| 2-Hydroxyethyl methacrylate + Choline formate ionic liquid | Stearic acid | Curcumin | N/A | Breast cancer (MCF-7 cells) | 16 |
| Combination agents delivery | |||||
| PLGA | DSPE-PEG | Doxorubicin, combretastatin A4 | N/A | Melanoma and Lewis lung carcinoma (B16F10 and Lewis lung carcinoma cells) | 22 |
| HPESO (hydrolyzed polymer of epoxidized soybean oil) | Tristearin–stearic acid 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 w/w :70 w/w |
Doxorubicin, Elacridar (GG918) | N/A | Multi-drug resistant breast cancer (MDA435/LCC6/MDR1 cells) breast cancer (EMT6/WT cells) | 32,71 |
| HPESO | Myristic acid | Doxorubicin, mitomycin C | N/A | Multi-drug resistant breast cancer (MDA-MB 435/LCC6/MDR1 cells) | 15 |
| PLGA | Soybean lecithin, DSPE-PEG | 2′-Deoxy-5-azacytidine (DAC), doxorubicin | N/A | Breast cancer (MDA-MB-231 cells) | 31 |
| PLA-PEG-PLA | PC, cholesterol, DSPE-PEG | TGF-β receptor-I inhibitor (SB505124), IL-2 | N/A | Melanoma (B16-F10 cells) | 79 |
| Diagnostic and theranostic agents delivery | |||||
| PLGA | Lecithin, DSPE-PEG | Iron oxide nanoparticles, camptothecin | N/A | Breast cancer (MT2 cells) | 33 |
| PLGA | DPPC, DSPE-PEG | Gold nanocrystals and paclitaxel in polymer core; sorafenib and Cy7 NIR dye in lipid shell | N/A | Colon cancer (LS174 T cells) | 86 |
| PLGA | Lecithin, DSPE-PEG | Doxorubicin, Indocyanin-green NIR dye | N/A | Basal cell carcinoma (BCC cells) | 54 |
| PLGA | Soybean lecithin | Gold nanocrystals, quantum dots | N/A | N/A | 14 |
| Targeted LPNs | |||||
| PLGA | Egg PC, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | 7 alpha-APTADD | Transferrin | Breast cancer (SKBR-3 cells) | 10 |
| PLGA | RBC vesicles | Anti-RBC IgG | Targets and neutralizes anti-RBC IgG | N/A | 88 |
| PLGA | RBC derived lipid membranes, FITC-PEG-lipid | DiD fluorescent dye | FITC, folate or AS1411 aptamer | Breast cancer (KB cells) | 89 |
| PLA | DSPE-PEG | Paclitaxel (PLA-conjugated) | KLWVLPK peptide for vascular endothelial targeting | N/A | 62 |
| PLGA | Lecithin, DSPE-PEG | Paclitaxel | AS1411 anti-nucleolin targeted aptamer | Breast cancer (MCF-7 cells) | 11 |
| Polyamidoamine grafted cholesterol | DOTAP:DOPE:cholesterol, DSPE-PEG |
Anti-EGFR siRNA | T7 peptide (HAIYPRH) to target transferrin | Breast cancer (MCF-7 cells) | 41 |
| PLGA | DSPE-PEG | 10-Hydroxycamptothecin | Cyclo(RGDyk) | Breast cancer (MCF-7 and MDA-MB-435s cells) | 12 |
| PLGA | DLPC, DSPE-PEG2K, DSPE-PEG5K | Docetaxel | Folate | Breast cancer (MCF-7 cells) | 17 |
| PLA | Soybean PC, DSPE-PEG, DSPE-PEG-FA | Mitomycin C | Folate | Lung cancer and hepatocellular carcinoma (A549 and H22 cells) | 34 |
| PLGA | DSPE-PEG-maleimide, lecithin | Doxorubicin | Anti-EGFR antibody | Hepatocellular carcinoma (SMMC-7721 cells) | 80 |
| PLGA | DSPE-PEG, lecithin | Paclitaxel | Half antibody of anti-CEA antibody | Pancreatic adenocarcinoma (BxPC3 cells) | 90 |
| PLGA | Lecithin, DSPE-PEG | Paclitaxel, combretastatin A4 | Cyclo(RGDFK) | Breast cancer (MCF-7 cells) | 61 |
| PLGA | PEG-OQLCS, cholesterol | Doxorubicin, pEGFP | Folate | Breast cancer (MDA-MB-231 cells) | 36 |
3.1. Targeting tumor cell proliferation
The delivery of cytotoxic agents to stall tumor cell proliferation has been the main focus of researchers working on LPNs. Many cytotoxic drugs have poor water solubility, low circulation half-life, and high non-specific toxicity. So far, LPNs have been used to deliver a variety of small molecule hydrophobic drugs, including docetaxel,17 salidroside,13 ginsenoside,29 paclitaxel,30 camptothecin,12 doxorubicin,27,31,32 sorafenib,14 and curcumin.16Using a single-step nanoprecipitation method, Zhang et al. developed LPNs for the delivery of docetaxel to prostate cancer cells.8 The nanoparticles were surface-conjugated with the A10 RNA aptamer to specifically target prostate cancer cells over-expressing the prostate specific membrane antigen (PSMA). The nanoparticles were shown to have high drug loading, sustained drug release over 120 h, and excellent stability.
Lee et al. synthesized LPNs that were loaded with ginsenoside Rg3, a pharmacologically active component of ginseng shown to have anti-cancer properties by inducing apoptosis and anti-angiogenesis.29 The nanoparticles were formulated using a solvent evaporation technique, and contained amphiphilic hyaluronic acid ceramide (HACE), PC and DSPE-PEG in the shell. They showed sustained release over a period of 16 days without any observed burst release. Blank nanoparticles were efficiently taken up by A549 cells through CD-44 mediated endocytosis, but had very minimal toxicity in vitro up to nanoparticle concentrations of 500 μg mL−1. The nanoparticles also showed a good circulation half-life profile in a Sprague–Dawley rat model.
Apart from more conventional LPNs, researchers have also developed stimuli-responsive LPNs to achieve greater control over drug delivery. In one such study, 10 nm iron oxide nanoparticles and camptothecin were encapsulated in the PLGA core, surrounded by soybean lecithin and DSPE-PEG in the shell.33 When the nanoparticles were triggered with a radio frequency (RF) magnetic field, localized heating in the nanoparticle core caused polymer degradation and drug release. The nanoparticles showed minimal drug release under control conditions, whereas with RF stimulation the drug release rate increased significantly, leading to 60% cell death.
Another good example of stimuli-responsive LPNs would be the study by Li et al., where they synthesized LPNs for the pH-triggered delivery of mitomycin C by reverse micelle–solvent evaporation.34 Mitomycin C and soybean phosphatidyl choline formed a complex in the core, and PLA surrounded the core. The micelles were further coated with a lipid layer containing DPPE, DSPE-PEG and DSPE-PEG-folic acid conjugates. A schematic illustrating the synthesis of the drug loaded LPNs is shown in Fig. 3, as well as the mechanism of folate targeting and drug release in the endo-lysosome. The nanoparticles exhibited a burst release in vitro, followed by a more sustained release over 120 h. They were taken up by A549 lung cancer cells within 4 h as shown by fluorescence microscopy and flow cytometry. In a xenograft mouse model, tumors treated with the drug-loaded nanoparticles shrank more than 50%, and folate-targeted nanoparticles accumulated better in tumors compared to non-targeted nanoparticles.
 | ||
| Fig. 3 pH-responsive LPNs. (A) Mitomycin C-loaded, folate-targeted LPNs for in vitro and in vivo targeted drug delivery. (B) An illustration of the sequence of events leading to cytotoxicity: tumor accumulation by the EPR effect, folate-receptor mediated cancer cell uptake, and subsequent release of mitomycin C in the endosome at low pH values. Reprinted with permission from ref. 34. Copyright © 2014 American Chemical Society. | ||
Researchers have also designed multi-functional LPNs to treat cancer more thoroughly. In an example of dual drug delivery, Su and coworkers synthesized LPNs for the co-delivery of epigenetic (2′-deoxy-5-azacytidine, DAC) and chemotherapeutic drugs (doxorubicin).31 DAC enhances the expression of tumor suppressor genes by inhibiting DNA methyltransferases that cause epigenetic silencing of these protective genes.35 DAC is also hypothesized to increase the sensitivity of cancer cells to doxorubicin. Using a nanoprecipitation method, both DAC and doxorubicin were encapsulated in the PLGA core and coated with a layer of lecithin and DSPE-PEG. The growth suppressive effects of the nanoparticles were tested on an MB231 human breast cancer cell line and synergistic growth inhibition was observed. In addition, HONE1 nasopharyngeal carcinoma cells treated with DAC nanoparticles showed better recovery in the expression of the tumor suppressor gene DLC1 (Deleted in Liver Cancer 1) compared to treatment with free drug.
Researchers have also explored the use of LPNs for the co-delivery of drugs and plasmids. Wang et al. designed LPNs with doxorubicin in the PLGA core and GFP-encoding plasmids complexed in the lipid shell, which were further coated with PEGylated amphiphilic octadecyl-quaternized lysine modified chitosan (PEG-OQLCS) and cationic folate acid coated amphiphilic octadecyl-quaternized lysine modified chitosan (FA-OQLCS).36 In comparison with uncoated PLGA nanoparticles and lipid-coated PLGA nanoparticles, lipid/folate-coated nanoparticles showed a more sustained release profile of doxorubicin over several days. The lipid/folate-coated nanoparticles were taken up in MDA-MB231 cells as shown by the intracellular fluorescence of both doxorubicin and GFP. Flow cytometry analysis showed that 46.6% of transfected cells had taken up both the plasmids and doxorubicin. The blank nanoparticles showed negligible toxicity whereas the drug loaded nanoparticles showed a dose-dependent toxicity that was slightly better than free drug.
Apart from inhibiting cell proliferation, other key aspects of cancer have also been exploited for LPN-based drug delivery. For example, researchers have explored the use of LPNs to deliver antisense RNA against growth-promoting oncogenes and tumor suppressor peptides, which are discussed in the following sections.
3.2. Targeting cancer-promoting oncogenes
Oncogenes are overexpressed by malignant cells and have always been a stalwart of cancer treatment strategies. One way to specifically downregulate oncogenes is to silence them using small interfering RNAs (siRNAs), a method that has led to promising therapy outcomes.37 But nucleic acids are highly charged and polyanionic, properties that do not favor cell penetration. In addition, their large molecular weights and rapid denaturation by nucleases in vivo necessitate the use of delivery vectors for efficient intracellular delivery.38 Researchers have recently become interested in delivering siRNAs in combination with other therapeutics to silence oncogenes.As a proof of concept for the silencing of genes involved in tumor metabolism, researchers delivered siRNA to knock down housekeeping genes such as glyceraldehyde 3-phosphate dehydrogenases (GAPDH). Shi et al. developed LPNs composed of 1,2-dimyristoleoyl-sn-glycero-3-ethylphosphocholine (DMOPC) and GAPDH siRNA in the PLGA core, and DSPE-PEG/lecithin mixtures in the shell.39 The nanoparticles were formed using a modified double emulsion/solvent evaporation technique with self-assembly. GAPDH siRNA delivered by LPNs to HepG2 hepatocytes resulted in efficient gene knockdown in vitro and at levels comparable to commercially available transfection reagents. In vivo efficacy studies in Balb/C nude mice showed GAPDH knockdown efficiencies of 42–45%.
The same group also reported the delivery of siRNA that targeted prohibitin (PHB1), a gene responsible for chemoresistance, cell proliferation and apoptosis.40 They synthesized LPNs from 1,2-epoxytetradecane with PAMAM dendrimers for the core and DSPE-PEG for the shell. PHB1 knockdown efficacy was tested in vitro in A549 cells and in vivo in an A549 xenograft mouse model. Nanoparticle treatment reduced cell proliferation significantly in vitro and reduced mouse tumor load by more than two-fold in vivo.
In a study that also included active targeting, Gao and coworkers developed and tested LPNs for siRNA delivery to cancer cells in vivo.41 The nanoparticle cores consisted of cholesterol-grafted poly(amidoamine) (rPAA-Chol polymer), while the shells contained DSPE-PEG modified with a peptide (T7-HAIYPRH) that targets the transferrin receptor overexpressed in cancer cells. The positive charge of the rPAA-Chol polymer enabled high siRNA loading via electrostatic interaction. LPN synthesis and cellular uptake pathways are illustrated in Fig. 4. Peptide targeting was shown to improve cellular uptake of the nanoparticles, as analyzed by fluorescence microscopy and flow cytometry. In MCF-7 cells, nanoparticles loaded with anti-EGFR siRNA showed 80% gene silencing in vitro, comparable to that when Lipofectamine 2000 is used. In an MCF-7 xenograft mouse model, the nanoparticles showed very low systemic toxicity, induced gene knockdown, and reduced the tumor load significantly.
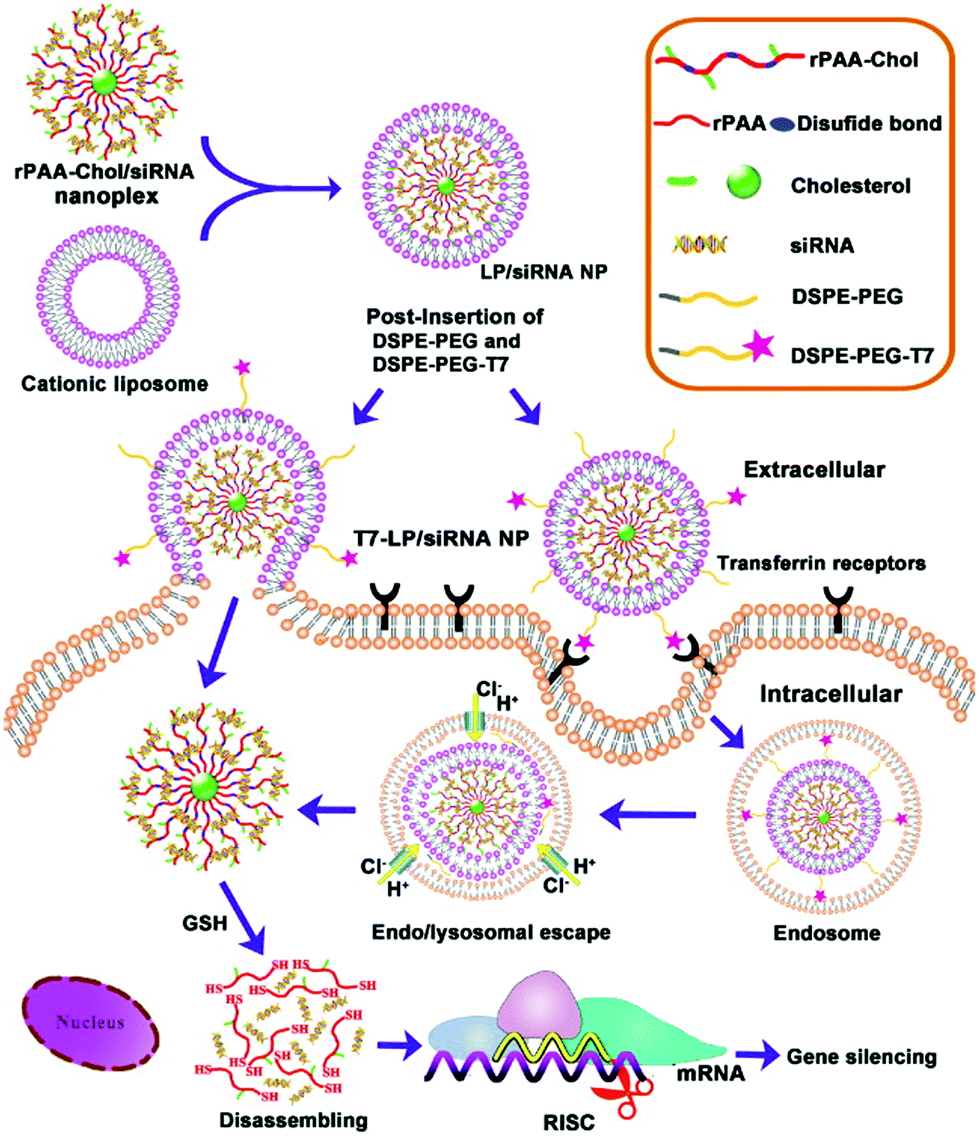 | ||
| Fig. 4 Schematic showing the two-step synthesis of lipid/rPAA-Chol LPNs (above) and LPN intracellular trafficking (below). Cellular targeting is mediated by the T7 peptide. Subsequent siRNA release from the disassembly of the nanoplex leads to silencing of the gene of interest. Reproduced with permission from ref. 41. Copyright © 2013, Elsevier Ltd. | ||
Using a unique technique called particle replication in nonwetting templates (PRINT), Hasan et al. developed highly monodisperse, uniformly sized LPNs.42 PRINT is a nano-molding technique that offers high control over size, shape and composition of the particles synthesized. A master template is first created using lithography, which is then loaded with the desired material via capillary filling. Uniform particles of desired characteristics can be peeled off from the mold using a harvesting film.43 In this study, PLGA nanoparticles were loaded with siRNA in the core and coated with DOTAP:DOPE in the shell. Interestingly, the nanoparticles were synthesized in a needle shape instead of the traditional spherical shape. Previous studies had shown that needle-shaped particles were more efficient at siRNA delivery than spherical-shaped particles, probably due to reduced uptake by the reticulo-endothelial system.44 The LPNs were used to deliver anti-Kinesin family member 11 (KIF11) siRNA, as inhibition of KIF11 is known to prevent centrosome migration and cause mitotic arrest and apoptosis in various cancers.45 The anti-cancer efficacy of the nanoparticles was tested in three prostate cancer cell lines, namely LNCaP, PC3 and DU145; in all cases, KIF11 knockdown and a reduction in cell viability were observed.
Yang et al. used a single step nanoprecipitation method to synthesize LPNs containing cationic lipid BHEM-Chol, the block polymer mPEG5k-PLA25k and the homopolymer PLA30k.24 The LPNs showed excellent loading of anti-Plk1 siRNA, good serum stability and cell internalization within 1 h. The nanoparticles were shown to escape from the endo-lysosome upon internalization, which is essential for post-transcriptional gene silencing in the cytoplasm. Polo-like kinase 1 (Plk1) was chosen for the role it plays in the maturation of the centrosome, making it a key regulator of cell division in eukaryotic cells. As the Plk1 oncogene overrides the regulatory function of p53, inhibition of Plk1 causes apoptosis in cancer cells by restoring p53 function.46 Anti-Plk1 siRNA nanoparticles inhibited mitotic progression and induced apoptosis in a BT474 xenograft mouse model, resulting in an almost five-fold reduction in tumor volume and a more than 60% knockdown in Plk1 gene expression.
In addition to intracellular drug targets, researchers have also found extracellular drug targets – the tumor microenvironment and tumor vasculature – to be highly attractive targets. The following sections describe studies that focus on these two targets, both of which are critical for tumor growth and metastasis.
3.3. Targeting the tumor microenvironment
The immediate niche of a tumor is called the tumor microenvironment. It consists of several types of non-malignant cells, an extracellular matrix and pre-existing and newly formed blood vessels, all of which are believed to support and promote tumor growth and facilitate metastasis.47 Evidence has shown that cancer cells frequently interact with their microenvironment, such as by recruiting leukocytes that secrete pro-proliferation and pro-angiogenic growth factors.48,49 Hence, aside from targeting the tumor itself, researchers have increasingly targeted the microenvironment to achieve a more effective therapeutic response.50Several features of the tumor microenvironment have been shown to be relevant for drug delivery using nanocarriers – namely the slightly elevated temperature, hypoxic and reductive environments, and leaky tumor vasculature.51 Apart from exploiting the inherent characteristics of the tumor microenvironment, various cell types and growth factors associated with the tumor microenvironment are also being targeted for effective tumor reduction. For example, tumor-associated macrophages and stromal cells are possible cellular targets,50 while the vasculature, the lymphatic system, inflammatory proteins and growth factors (such as PDGF, VEGF and HGF) are also possible molecular targets.50
Photothermal therapy52 and photodynamic therapy53 have been employed to effectively ablate the tumor microenvironment with some successful studies reported. To ablate the tumor as well as cells in the surrounding microenvironment, Zheng et al. developed LPNs containing doxorubicin and indocyanine green (ICG).54 Using a one-step sonication method, doxorubicin and ICG were loaded into the PLGA core, and lecithin and DSPE-PEG were self-assembled over the core. ICG has been approved by the FDA for human use, and can be used to convert absorbed light into heat for photothermal applications.55 In addition, its NIR emission around 800 nm can be used for excellent deep tissue imaging due to low background autofluorescence. In this study, 8 min of NIR laser irradiation raised the temperature of the solution from room temperature to 53.2 degrees, which is sufficient to cause irreversible damage to the cancer cells. In addition, irradiation led to thermally-induced disruption of the nanoparticles and doxorubicin release. In both MCF-7 (doxorubicin-sensitive) and MCF-7/ADR (doxorubicin-resistant) cell lines, the combination of photothermal therapy and chemotherapy was synergistic and resulted in significant cell death. Biodistribution studies performed in an MCF-7 xenograft mouse model showed tumor accumulation of the nanoparticles. A single nanoparticle dose combined with laser irradiation led to complete remission for both doxorubicin-sensitive and doxorubicin-resistant tumors, with no recurrence observed during the 30 day study.
A study by Clawson et al. highlighted a pH-sensitive LPN system that exploits the acidic nature of the tumor microenvironment.28 The nanoparticles had a pH-sensitive lipid-(succinate)-mPEG shell that is stable at physiological pH but not at lower pH values. In the acidic tumor microenvironment, the lipid-(succinate)-mPEG underwent acid hydrolysis, allowing the PEG layer to be hydrolysed away. Once the PEG shell was shed, the exposed fusogenic lipids on the nanoparticle surface facilitated the aggregation of nanoparticles by forming multilamellar lipid structures, as tested in buffers with pH values ranging from 7.4 to 3. Although not shown here, the PLGA core could be used to encapsulate hydrophobic drugs for tumor-specific delivery.
In an example of pH-assisted gene delivery, Su et al. used a double emulsion method to design LPNs. The LPNs had a reduction-sensitive core of poly(β-amino ester) (PBAE), enveloped by a phospholipid bilayer shell consisting of DOPC, DOTAP and DSPE-PEG.56 The nanoparticles exhibited rapid dissolution in acidic solutions compared to pH-insensitive LPNs. Synthetic double-stranded polyriboinosinic-polyribocytidylic acid (poly I/C) mRNA was delivered by loading onto the LPN surface via electrostatic interaction. Poly I/C was chosen as it is an effective inducer of interferon and has been shown to have anti-neoplastic properties in several human cancers.57 Poly I/C loaded LPNs exhibited successful endosomal escape and antigen presentation in dendritic DC2.4 cells. The researchers also demonstrated the successful delivery of luciferase-loaded LPNs, which were delivered via intranasal administration in a C57BL/6J mouse model.
3.4. Targeting the tumor vasculature
Establishing a functional vascular supply is vital for the growth and development of solid tumors. Tumor-associated blood vessels are structurally and functionally different from normal vasculature, making the targeting of angiogenesis in tumors a promising option for cancer therapy.58Targeting the tumor vasculature presents a new challenge altogether, since shutting down the tumor vasculature hinders delivery of therapeutics into the tumor and also increases hypoxia, which has been correlated with enhanced resistance and metastasis.59,60 To circumvent these issues, Sengupta et al. developed a LPN system that used a sequential drug release strategy: the nanoparticles first released combretastatin A-4, an anti-angiogenic factor that targets neovasculature, from the shell, before releasing doxorubicin from the core.22 The nanoparticles were shown to shut down the vasculature in an in vitro 3D coculture of endothelial cells, trapping the nanoparticles locally in the tumor microenvironment. The nanoparticles then released doxorubicin in a sustained manner from the core, ablating melanoma cells in culture. The nanoparticles were tested in C57/BL6 mice bearing melanoma or Lewis lung carcinoma tumor xenografts. In survival studies, dual drug delivery led to superior tumor suppression in comparison with treatment with either therapeutic delivered alone.
Wang et al. used a sequential drug release strategy similar to Sengupta et al., but incorporated additional vascular targeting factors for active targeting.61 Paclitaxel was loaded into the PLGA core, while combretastatin A4 was incorporated into a lipid monolayer containing lecithin, distearoyl-sn-glycero-phosphocholine (DSPC), cholesterol and RGDfK peptide-conjugated DSPE-PEG. Here, RGD targeting peptides were added as they are known to preferentially bind to the αvβ3 integrin expressed on tumor endothelial cells. The nanoparticles were tested in an MCF-7 breast cancer cell line, where they induced significant apoptosis; and also in HUVEC cells, where the cells took up about 90% of the targeted nanoparticles within 1 h in comparison with 20% for non-targeted nanoparticles. Temporal release and sequential eradication of vasculature and the tumor cells were observed in a 3D co-culture model of endothelial and cancer cells.
Chan et al. designed a hybrid nanoparticle system, called nanoburrs, to target components of the extracellular matrix.62 The nanoparticles were synthesized by nanoprecipitation and had a polymeric core (PLA) and a lipid shell (soybean lecithin and DSPE-PEG). The drug of choice, paclitaxel, was conjugated to PLA so that it would be released gradually by ester hydrolysis. The nanoparticles were surface-functionalized with a peptide against collagen IV – collagen IV is found in the basement membrane underlying the endothelial monolayer and plays a major role in angiogenesis, cancer cell growth, adhesion, migration and differentiation.63In vitro, the nanoburrs showed a sustained release of paclitaxel over 12 days and reduced the proliferation of human aortic smooth muscle cells. In ex vivo and in vivo rat injury models, the nanoparticles were found to accumulate at significantly higher amounts in angioplastied arteries as compared to healthy arteries. Even though the nanoburrs were studied in the context of cardiovascular disease, they may potentially be used to target the tumor vasculature.
Despite advancements in the treatment of cancer, two major hurdles persist – the development of multi-drug resistance and metastasis. Nanomedicine may offer some relief to traditional chemotherapy approaches, as discussed in the following two sections.
3.5. Targeting multi-drug resistance
Acquiring multi-drug resistance is a major hallmark of many cancers and poses a big challenge to chemotherapy.64 Selection pressures from the microenvironment drive the accumulation of mutations in cancer cells, leading to phenotypic changes such as the overexpression of drug-effluxing protein pumps.65 Clinically, the presence of multi-drug resistance in patient tumor biopsies has been correlated to poor prognosis.66,67 The delivery of drugs using nanoparticles has been shown to circumvent multi-drug resistance by lowering the apoptotic threshold of cancer cells.68Two or more drugs that are used together can cause additive, synergistic, potentiated or antagonistic effects.69 By delivering multiple agents with favorable interactions, researchers can target different pathways at the same time, discouraging the development of resistance and achieving better treatment outcomes compared to single agent therapy.70
In one such example of dual drug delivery, Wong and coworkers developed LPNs for the co-delivery of a chemotherapeutic drug (doxorubicin) and a chemosensitizer (Elacridar-GG918).71 GG918, a new generation of P-glycoprotein (P-gp) inhibitors with better potency and lower toxicity, works by preventing the efflux of doxorubicin from cells. The nanoparticles were prepared by ultrasonication using a HPESO (hydrolyzed polymer of epoxidized soybean oil) core and a tristearin–stearic acid (30:70) lipid shell. The nanoparticles were synthesized in the size range of 150–270 nm and had excellent drug encapsulation efficiencies. In a drug resistant cell line, MDA435/LCC6/MDR1, combination nanoparticles showed two-fold higher toxicity in comparison with free drug combinations, as GG918 prevented doxorubicin from being effluxed. Finally, the long-term growth suppressive effect of the LPNs was tested in a clonogenic assay. The combination drug loaded nanoparticles showed the lowest IC50 of 0.34 μg mL−1 and inhibited colony formation.
Aside from delivering a chemosensitizer to overcome resistance, researchers have also exploited the ability of nanoparticles to deliver high concentrations of drugs into cancer cells to overcome resistance. The same group has earlier compared doxorubicin-loaded LPNs against doxorubicin-conjugated HPESO polymer nanoparticles.72 The LPNs showed greater efficacy in vitro compared to HPESO nanoparticles, and were very effective in overcoming resistance in P-gp over-expressing MDA435/LCC6 cells and EMT6 cells. The LPNs were observed to enter the cells via phagocytosis. Concomitantly, the LPNs released the drugs into the extracellular space, which were then taken up by the cells by simple diffusion.
In an attempt to address multi-drug resistance, Zhang et al. developed an LPN system for the delivery of mitoxantrone, a water-soluble cationic drug that is a substrate of P-gp efflux pumps.73 A negatively charged dextran sulfate sodium polymer was used in the core to maximize drug loading via electrostatic complexation. The lipid shell was formed using Compritol 888 ATO, Cremophor RH40, Miglyol 812 and lecithin using an ultrasonication–emulsification method. The nanoparticles were highly effective in reducing the viability of both drug sensitive MCF-7 cells and drug-resistant, breast cancer resistant protein (BCRP)-overexpressing MCF-7/MX cells. Mitoxantrone delivery also increased the sensitivity of resistant cells to drugs by up to 4.6-fold. Mechanistically, it was demonstrated that mitoxantrone-loaded nanoparticles entered the cells via clathrin-mediated endocytosis and overcame resistance by escaping BCRP transporter-induced efflux.
Another strategy adopted by Prasad et al. to overcome multi-drug resistance is to co-deliver doxorubicin and mitomycin C.15 The nanoparticles contained HPESO in the core, and contained myristic acid, PEG40SA and PEG100SA in the lipid monolayer. The nanoparticles showed good in vitro cytotoxicity in MDA-MB 435/LCC6/WT cells and in P-gp overexpressing MDA-MB 435/LCC6/MDR1 cells. In an orthotopic human breast cancer mouse model, treatment with the nanoparticles led to a 108–151% increase in tumor growth delay, a 210–316% increase in life span, and a 10–20% de facto cure after treatment. Treated mice did not exhibit any significant loss in body weight.
3.6. Targeting tumor metastasis
Metastasis presents the single most significant challenge in cancer management, being the leading cause of treatment failures.74 About 90% of cancer deaths are due to metastatic tumors rather than primary tumors. Metastasis encompasses a complex sequence of events starting with circulating tumor cells dislodged from a primary tumor. Subsequently, the circulating tumor cells adhere to vascular walls at a secondary site and migrate across the endothelium to establish a secondary tumor.75 Recently, researchers have identified an inherently resistant sub-population of tumor cells, termed cancer stem cells (CSCs), which play a predominant role in tumor recurrence and metastasis.76 Here, we explore studies that use LPNs to target CSCs or metastatic cancers.Palange et al. showed that by reducing vascular inflammation, researchers can significantly prevent the metastasis of primary tumors.23 In this study, they developed NANOCurc nanoparticles that contain curcumin in the PLGA core and DPPC and DSPE-PEG in the shell. Curcumin, the active component in the turmeric spice, is being increasingly explored as an anti-cancer agent owing to its anti-inflammatory properties, one being the inhibition of nitric oxide.77 Curcumin-loaded LPNs were shown to down-regulate cell adhesion molecules such as ICAM-1 and MUC-1 within 1 h of treatment. By downregulating important cell-surface proteins, vascular inflammation was reduced and the movement of circulating tumor cells along the vasculature was impaired, thereby preventing metastasis (Fig. 5). Time- and dose-dependent toxicity was also observed in MDA-MB231 and HUVEC cells treated with NANOCurc nanoparticles.
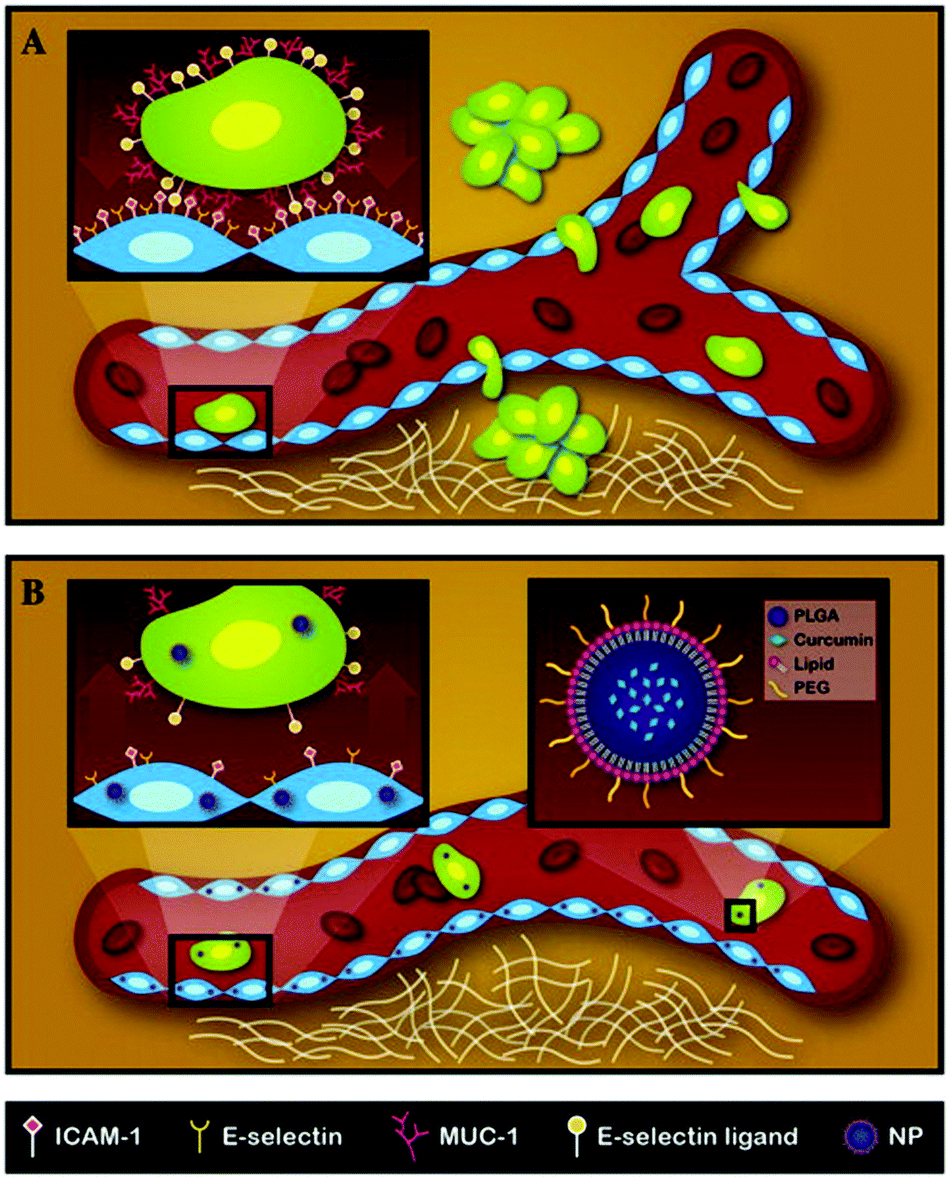 | ||
| Fig. 5 Schematic representation of the metastatic process and mechanism of action for NANOCurc. (A) Circulating tumor cells metastasize from the primary site and form tumors at a secondary site. The inset shows the interaction of a tumor cell with endothelial cells through adhesion molecules. (B) NANOCurc treatment inhibits metastasis by reducing the expression of adhesion molecules on endothelial cells. Reproduced with permission from ref. 23. Copyright © 2014, Elsevier Inc. | ||
Conventional immunotherapy has been used to target metastatic cancers. The over-expression of certain growth factors (e.g. TGF-β) by the tumor and its microenvironment, however, impedes the effectiveness of such treatments, as these growth factors lead to growth deregulation, angiogenesis and immunosuppressive effects.78 To overcome the inhibitory effects caused by TGF-β, Park et al. developed liposomal polymeric gels loaded with a TGF-β inhibitor (SB505124) and an immunity-boosting cytokine (IL-2). SB505124 was initially conjugated to β-cyclodextrin, after which the nanoparticles were formed with PC, DSPE-PEG, cholesterol and IL-2 using an extrusion/freeze drying method. It was observed that encapsulation of the drugs in the nanoparticles decreased drug clearance and improved biodistribution in vivo in a mouse model of melanoma. The co-loaded nanoparticles showed excellent inhibitory effects in comparison with nanoparticle-based monotherapies. The combination treatment also resulted in a greater than 20% increase (per gram of tumor mass) in activated CD8+ T lymphocytes compared to untreated mice, without changing the overall CD4/CD8 ratio and regulatory T cell numbers. An increase in the number of natural killer cells per gram of tumor mass was also observed as a result of combination therapy. Thus, apart from tumor reduction, the particles also activated innate and adaptive immune responses.79
To target the stem cell population in tumors, Gao et al. synthesized LPNs by a single step nanoprecipitation method with doxorubicin and PLGA in the core and soybean lecithin and DSPE-PEG-maleimide in the shell.80 In addition, the nanoparticles were surface-conjugated with anti-EGFR antibodies to target hepatocellular carcinoma cells. The nanoparticles induced significant toxicity and were taken up by different hepatocellular carcinoma cell lines (SMMC-7721, HepG2 and Huh7) very efficiently. In flow cytometry studies, the percentage of side population cells was found to be reduced by half, indicating that the LPNs could target CSCs. Finally, in vivo efficacy studies in an SMMC-7721 HCC xenograft mouse model showed a five-fold reduction in tumor load in comparison with non-targeted nanoparticles and free adriamycin.
Researchers have also developed LPNs for combinatorial chemotherapy and radiotherapy. Here, Werner et al. delivered paclitaxel and yttrium-90 (90Y) for the treatment of ovarian cancer peritoneal metastasis.81 The ChemoRad nanoparticles had a PLGA core containing paclitaxel and 90Y, and a shell consisting of DSPE-PEG-folate, lecithin and DMPE-DTPA. The folate-targeted nanoparticles were taken up more efficiently by folate receptor-overexpressing SKOV3 cells in vitro, as compared to SW626 cells which do not overexpress the folate receptor. Clonogenic assays carried out with SKOV3 and OVCAR3 cells showed higher in vitro cytotoxicity by targeted NPs compared to non-targeted NPs. In vivo evaluation of the targeted ChemoRad NPs in a murine model of ovarian cancer peritoneal metastasis showed prolonged survival times and a reduction in tumor growth, while no statistical significance could be reached when either therapeutic was used alone.
Given the versatility of nanoparticulate systems, it is possible to incorporate additional functionalities such as imaging into these drug carriers. The ability to track the biodistribution of LPNs in vivo is highly beneficial in clinical cancer management, making highly attractive nanoparticles that can perform dual roles of drug delivery and imaging. Such LPNs are described in the following section.
3.7. Theranostic approaches in cancer treatment
According to the American Cancer Society, patient survival rates for those treated with early stage cancers are much higher (>90%) than at advanced or metastatic stages, indicating the importance of diagnosis in cancer treatment. But the diagnosis of a cancer is not always straightforward, and there exist many hurdles in using imaging agents in vivo. For example, there are several intrinsic disadvantages with traditional fluorophores and contrast agents, and these include low selectivity and sensitivity, photobleaching, short circulation times and toxicity issues.82Recent developments in nanotechnology have enabled the use of nanoparticles for imaging.83 Typically, the fluorescent dye or contrast agent is encapsulated in the nanoparticles to overcome their solubility and toxicity issues. Targeting agents such as antibodies and aptamers can be conjugated onto the surface of these nanoparticles, while therapeutic agents can be encapsulated into the core. The ability to combine diagnostics and therapy, or ‘theranostics’, makes nanoparticles an invaluable asset in the clinical toolkit.
In one such example, Aryal et al. developed LPNs for magnetic resonance imaging (MRI) using gadolinium ions and iron oxide nanoparticles.84 The core of the nanoparticle contained PLGA and ultra-small superparamagnetic iron oxide nanoparticles (USPIO), while the shell contained lipid-PEG and DOTA chelated to gadolinium ions. The nanoparticles were uniformly sized, monodisperse and showed good contrast agent loading. They also had significantly higher magnetic relaxivities in comparison with commercially available contrast agents, and showed efficient uptake by J-774 cells in vitro. In a murine B16-F10 xenograft model, the nanoparticles accumulated in the tumor within 3 h and showed excellent contrast. Here, drug loading was not demonstrated although the PLGA core could be used to encapsulate drugs.
Aside from MRI imaging, researchers have also used gold nanoparticles for computed tomography (CT) imaging. In a study by Willem et al., gold nanoparticles and quantum dots were chemically conjugated to the PLGA core and coated with soybean lecithin and DSPE-PEG.14 The gold nanoparticles were useful for CT imaging and the quantum dots were useful for fluorescence imaging, as tested in J-774A.1 mouse macrophage cells in vitro. Once again, the PLGA core could be used to encapsulate therapeutic molecules but the researchers did not demonstrate so in their study.
Liao et al. developed LPNs for simultaneous imaging and drug delivery.85 Doxorubicin was loaded into the PLGA core, after which it was coated with a paramagnetic lipid shell consisting of amphiphilic octadecyl-quaternized lysine-modified chitosan (OQLCS) individually conjugated to PEG, Gd-DTPA and folic acid by gentle hydration. Here, the nanoparticles were modified with folic acid for cancer-specific targeting and gadolinium for contrast enhancement. In in vitro studies, the nanoparticles showed good stability and better contrast than commercially available contrast agents such as Magnevist®. The nanoparticles also loaded doxorubicin efficiently and were taken up by HeLa cells within 4 h. The cytotoxic effect of the drug-loaded nanoparticles was slightly lower than with free doxorubicin but was comparable.
In another example of theranostic application of LPNs, Mieszawska et al. used a microfluidic technique to synthesize LPNs. The LPNs consisted of PLGA, gold nanoparticles (for CT imaging) and doxorubicin (hydrophobic drug) in the core followed by sorafenib (lipophilic drug) and Cy7 (for near infrared imaging) in the shell.86 The nanoparticles exhibited controlled drug release over 20 days, and inhibited growth in LS174 T cancer cell lines. The biodistribution of the nanoparticles was studied in a LS174 T xenograft mouse model. Both 3D CT imaging and near infrared fluorescence imaging showed nanoparticle accumulation to be significant only in the tumor 18 h post injection.
4. Conclusions and future directions
The last decade has seen interesting developments in the field of lipid–polymer hybrid nanotechnology. There has been an exponential increase in the number of studies performed using these hybrid systems, with a dazzling array of surface coatings and encapsulated cargoes. Not only have these LPNs been used to deliver therapeutic molecules such as small molecules, plasmids, siRNA, proteins and peptides, they have also been used to deliver diagnostic molecules such as iron oxide nanoparticles, fluorescent dyes, quantum dots and gold nanoparticles. These systems have been tested in both in vitro and in vivo models.Although LPNs encompass the advantages of both polymeric nanoparticles and liposomes, they present new challenges that may impede clinical translation. For example, a multi-step synthesis process is required due to the use of two classes of biomaterials (lipids and polymers), which may make scale-up manufacturing more challenging. There are also additional economic and quality considerations when producing the LPNs on a large scale. Researchers need to define all of the component ingredients at a molecular level, and evaluate their safety and biocompatibility in vivo. Thus, the development of simple and scalable protocols for the synthesis of LPNs, using FDA-approved materials with highly predictable in vivo characteristics, will enable faster clinical translation.
Although LPNs present a number of advantages in that they can deliver both hydrophobic and hydrophilic cargo and also be functionalized for active targeting, more studies need to be done at the pre-clinical stage. For example, the majority of cancer therapeutic studies reported in the literature using LPNs have only been tested in in vitro models. More investigational new drug (IND) type studies focusing on in vivo antitumor efficacy, biodistribution, clearance and long-term toxicity will need to be performed.
Apart from IND studies, there are also several interesting novel techniques that could be explored further. One such example is the recent development of ‘cell ghosts’, which are LPN-like nanoparticles that use biologically derived lipid membranes isolated from cells in place of synthetic lipids.87 Such nanoparticles may have unique biological functionalities depending upon the cell type chosen and hold immense potential for cancer treatment. For example, coating a nanoparticle with biological cell membranes bearing self-antigens will prolong their circulation half-life; these particles may more effectively evade the reticulo-endothelial system compared to synthetic stealth materials such as PEG. Further, coating nanoparticles with biological membranes already having the right display of cell-surface ligands can lead to more precise targeting, compared with transfection and expression of the cell-surface ligand using genetic modification techniques. While cell ghost technology is very appealing given its potential, several factors like cross-immunogenicity, uniformity and feasibility of large scale synthesis need to be investigated in detail.
With more pre-clinical studies (and possibly clinical studies) foreseen to take place over the next few years, we believe that more encouraging data will be reported that demonstrate the versatility of LPNs as a drug delivery carrier and a platform of choice for cancer drug delivery.
Acknowledgements
JMC acknowledges start-up grant funding from Nanyang Technological University (NTU) and the Lee Kong Chian School of Medicine (LKCMedicine).References
- S. S. Lim, T. Vos, A. D. Flaxman, G. Danaei, K. Shibuya, H. Adair-Rohani, M. A. AlMazroa, M. Amann, H. R. Anderson, K. G. Andrews, M. Aryee, C. Atkinson, L. J. Bacchus, A. N. Bahalim, K. Balakrishnan, J. Balmes, S. Barker-Collo, A. Baxter, M. L. Bell, J. D. Blore, F. Blyth, C. Bonner, G. Borges, R. Bourne, M. Boussinesq, M. Brauer, P. Brooks, N. G. Bruce, B. Brunekreef, C. Bryan-Hancock, C. Bucello, R. Buchbinder, F. Bull, R. T. Burnett, T. E. Byers, B. Calabria, J. Carapetis, E. Carnahan, Z. Chafe, F. Charlson, H. Chen, J. S. Chen, A. T.-A. Cheng, J. C. Child, A. Cohen, K. E. Colson, B. C. Cowie, S. Darby, S. Darling, A. Davis, L. Degenhardt, F. Dentener, D. C. Des Jarlais, K. Devries, M. Dherani, E. L. Ding, E. R. Dorsey, T. Driscoll, K. Edmond, S. E. Ali, R. E. Engell, P. J. Erwin, S. Fahimi, G. Falder, F. Farzadfar, A. Ferrari, M. M. Finucane, S. Flaxman, F. G. R. Fowkes, G. Freedman, M. K. Freeman, E. Gakidou, S. Ghosh, E. Giovannucci, G. Gmel, K. Graham, R. Grainger, B. Grant, D. Gunnell, H. R. Gutierrez, W. Hall, H. W. Hoek, A. Hogan, H. D. Hosgood III, D. Hoy, H. Hu, B. J. Hubbell, S. J. Hutchings, S. E. Ibeanusi, G. L. Jacklyn, R. Jasrasaria, J. B. Jonas, H. Kan, J. A. Kanis, N. Kassebaum, N. Kawakami, Y.-H. Khang, S. Khatibzadeh, J.-P. Khoo, C. Kok, F. Laden, R. Lalloo, Q. Lan, T. Lathlean, J. L. Leasher, J. Leigh, Y. Li, J. K. Lin, S. E. Lipshultz, S. London, R. Lozano, Y. Lu, J. Mak, R. Malekzadeh, L. Mallinger, W. Marcenes, L. March, R. Marks, R. Martin, P. McGale, J. McGrath, S. Mehta, Z. A. Memish, G. A. Mensah, T. R. Merriman, R. Micha, C. Michaud, V. Mishra, K. M. Hanafiah, A. A. Mokdad, L. Morawska, D. Mozaffarian, T. Murphy, M. Naghavi, B. Neal, P. K. Nelson, J. M. Nolla, R. Norman, C. Olives, S. B. Omer, J. Orchard, R. Osborne, B. Ostro, A. Page, K. D. Pandey, C. D. H. Parry, E. Passmore, J. Patra, N. Pearce, P. M. Pelizzari, M. Petzold, M. R. Phillips, D. Pope, C. A. Pope III, J. Powles, M. Rao, H. Razavi, E. A. Rehfuess, J. T. Rehm, B. Ritz, F. P. Rivara, T. Roberts, C. Robinson, J. A. Rodriguez-Portales, I. Romieu, R. Room, L. C. Rosenfeld, A. Roy, L. Rushton, J. A. Salomon, U. Sampson, L. Sanchez-Riera, E. Sanman, A. Sapkota, S. Seedat, P. Shi, K. Shield, R. Shivakoti, G. M. Singh, D. A. Sleet, E. Smith, K. R. Smith, N. J. C. Stapelberg, K. Steenland, H. Stöckl, L. J. Stovner, K. Straif, L. Straney, G. D. Thurston, J. H. Tran, R. Van Dingenen, A. van Donkelaar, J. L. Veerman, L. Vijayakumar, R. Weintraub, M. M. Weissman, R. A. White, H. Whiteford, S. T. Wiersma, J. D. Wilkinson, H. C. Williams, W. Williams, N. Wilson, A. D. Woolf, P. Yip, J. M. Zielinski, A. D. Lopez, C. J. L. Murray and M. Ezzati, Lancet, 2012, 380, 2224–2260 CrossRef.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- L. Zhang, F. Gu, J. Chan, A. Wang, R. Langer and O. Farokhzad, Clin. Pharmacol. Ther., 2007, 83, 761–769 CrossRef PubMed.
- A. Sharma and U. S. Sharma, Int. J. Pharm., 1997, 154, 123–140 CrossRef CAS.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149 CrossRef CAS PubMed.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS.
- I. De Miguel, L. Imbertie, V. Rieumajou, M. Major, R. Kravtzoff and D. Betbeder, Pharm. Res., 2000, 17, 817–824 CrossRef CAS.
- L. Zhang, J. M. Chan, F. X. Gu, J.-W. Rhee, A. Z. Wang, A. F. Radovic-Moreno, F. Alexis, R. Langer and O. C. Farokhzad, ACS Nano, 2008, 2, 1696–1702 CrossRef CAS PubMed.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 1997, 28, 5–24 CrossRef CAS.
- Y. Zheng, B. Yu, W. Weecharangsan, L. Piao, M. Darby, Y. Mao, R. Koynova, X. Yang, H. Li and S. Xu, Int. J. Pharm., 2010, 390, 234–241 CrossRef CAS PubMed.
- A. Aravind, P. Jeyamohan, R. Nair, S. Veeranarayanan, Y. Nagaoka, Y. Yoshida, T. Maekawa and D. S. Kumar, Biotechnol. Bioeng., 2012, 109, 2920–2931 CrossRef CAS PubMed.
- Z. Yang, X. Luo, X. Zhang, J. Liu and Q. Jiang, Biomed. Mater., 2013, 8, 025012 CrossRef PubMed.
- D.-L. Fang, Y. Chen, B. Xu, K. Ren, Z.-Y. He, L.-L. He, Y. Lei, C.-M. Fan and X.-R. Song, Int. J. Mol. Sci., 2014, 15, 3373–3388 CrossRef CAS PubMed.
- J. Willem, Chem. Commun., 2012, 48, 5835–5837 RSC.
- P. Prasad, A. Shuhendler, P. Cai, A. M. Rauth and X. Y. Wu, Cancer Lett., 2013, 334, 263–273 CrossRef CAS PubMed.
- S. S. D. Kumar, A. Mahesh, S. Mahadevan and A. B. Mandal, Biochim. Biophys. Acta, 2014, 1840, 1913–1922 CrossRef CAS PubMed.
- Y. Liu, K. Li, J. Pan, B. Liu and S.-S. Feng, Biomaterials, 2010, 31, 330–338 CrossRef CAS PubMed.
- P. Zhao, H. Wang, M. Yu, Z. Liao, X. Wang, F. Zhang, W. Ji, B. Wu, J. Han and H. Zhang, Eur. J. Pharm. Biopharm., 2012, 81, 248–256 CrossRef CAS PubMed.
- J. Thevenot, A.-L. Troutier, L. David, T. Delair and C. Ladavière, Biomacromolecules, 2007, 8, 3651–3660 CrossRef CAS PubMed.
- A.-L. Troutier, T. Delair, C. Pichot and C. Ladavière, Langmuir, 2005, 21, 1305–1313 CrossRef CAS PubMed.
- A.-L. Troutier, L. Véron, T. Delair, C. Pichot and C. Ladavière, Langmuir, 2005, 21, 9901–9910 CrossRef CAS PubMed.
- S. Sengupta, D. Eavarone, I. Capila, G. Zhao, N. Watson, T. Kiziltepe and R. Sasisekharan, Nature, 2005, 436, 568–572 CrossRef CAS PubMed.
- A. L. Palange, D. Di Mascolo, C. Carallo, A. Gnasso and P. Decuzzi, Nanomedicine, 2014, 10, 991–1002 CrossRef CAS PubMed.
- X.-Z. Yang, S. Dou, Y.-C. Wang, H.-Y. Long, M.-H. Xiong, C.-Q. Mao, Y.-D. Yao and J. Wang, ACS Nano, 2012, 6, 4955–4965 CrossRef CAS PubMed.
- F. Huang, M. You, T. Chen, G. Zhu, H. Liang and W. Tan, Chem. Commun., 2014, 50, 3103–3105 RSC.
- L. Zhang and L. Zhang, Nano Life, 2010, 1, 163–173 CrossRef CAS.
- X. Dong, C. A. Mattingly, M. T. Tseng, M. J. Cho, Y. Liu, V. R. Adams and R. J. Mumper, Cancer Res., 2009, 69, 3918–3926 CrossRef CAS PubMed.
- C. Clawson, L. Ton, S. Aryal, V. Fu, S. Esener and L. Zhang, Langmuir, 2011, 27, 10556–10561 CrossRef CAS PubMed.
- J. Y. Lee, H. Yang, I. S. Yoon, S. B. Kim, S. H. Ko, J. S. Shim, S. H. Sung, H. J. Cho and D. D. Kim, J. Pharm. Sci., 2014, 103, 3254–3262 CrossRef CAS PubMed.
- Y. Liu, J. Pan and S.-S. Feng, Int. J. Pharm., 2010, 395, 243–250 CrossRef CAS PubMed.
- X. Su, Z. Wang, L. Li, M. Zheng, C. Zheng, P. Gong, P. Zhao, Y. Ma, Q. Tao and L. Cai, Mol. Pharm., 2013, 10, 1901–1909 CrossRef CAS PubMed.
- H. L. Wong, A. M. Rauth, R. Bendayan and X. Y. Wu, Eur. J. Pharm. Biopharm., 2007, 65, 300–308 CrossRef CAS PubMed.
- S. Deok Kong, M. Sartor, C.-M. Jack Hu, W. Zhang, L. Zhang and S. Jin, Acta Biomaterialia, 2013, 9, 5447–5452 CrossRef CAS PubMed.
- Y. Li, H. Wu, X. Yang, M. Jia, Y. Li, Y. Huang, J. Lin, S. Wu and Z. Hou, Mol. Pharm., 2014, 11, 2915–2927 CrossRef CAS PubMed.
- G. Reid, J. Besterman and A. MacLeod, Curr. Opin. Mol. Ther., 2002, 4, 130–137 CAS.
- H. Wang, P. Zhao, W. Su, S. Wang, Z. Liao, R. Niu and J. Chang, Biomaterials, 2010, 31, 8741–8748 CrossRef CAS PubMed.
- O. Heidenreich, in siRNA and miRNA Gene Silencing, Springer, 2009, pp. 1–22 Search PubMed.
- E. Mastrobattista, W. E. Hennink and R. M. Schiffelers, Pharm. Res., 2007, 24, 1561–1563 CrossRef CAS PubMed.
- J. Shi, Z. Xiao, A. R. Votruba, C. Vilos and O. C. Farokhzad, Angew. Chem., Int. Ed., 2011, 123, 7165–7169 CrossRef.
- J. Shi, Y. Xu, X. Xu, X. Zhu, E. Pridgen, J. Wu, A. R. Votruba, A. Swami, B. R. Zetter and O. C. Farokhzad, Nanomedicine, 2014, 10, 897–900 CrossRef CAS PubMed.
- L.-Y. Gao, X.-Y. Liu, C.-J. Chen, J.-C. Wang, Q. Feng, M.-Z. Yu, X.-F. Ma, X.-W. Pei, Y.-J. Niu and C. Qiu, Biomaterials, 2014, 35, 2066–2078 CrossRef CAS PubMed.
- W. Hasan, K. Chu, A. Gullapalli, S. S. Dunn, E. M. Enlow, J. C. Luft, S. Tian, M. E. Napier, P. D. Pohlhaus and J. P. Rolland, Nano Lett., 2011, 12, 287–292 CrossRef PubMed.
- J. Desimone, J. Wang and Y. Wang, Janus Part. Synth. Self-Assem. Appl., 2012, 1, 90–107 CAS.
- P. Kolhar, N. Doshi and S. Mitragotri, Small, 2011, 7, 2094–2100 CrossRef CAS PubMed.
- N. P. Ferenz, A. Gable and P. Wadsworth, Seminars in cell & developmental biology, 2010 Search PubMed.
- X. Liu and R. L. Erikson, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5789–5794 CrossRef CAS PubMed.
- J. A. Joyce and J. W. Pollard, Nat. Rev. Cancer, 2008, 9, 239–252 CrossRef PubMed.
- A. Alphonso and S. K. Alahari, Neoplasia, 2009, 11, 1264–1271 CAS.
- T. Ishimoto, H. Sawayama, H. Sugihara and H. Baba, J. Gastroenterol., 2014, 1–10 Search PubMed.
- J. A. Joyce, Cancer Cell, 2005, 7, 513–520 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- A. M. Gobin, J. J. Moon and J. L. West, Int. J. Nanomed., 2008, 3, 351 CAS.
- C. J. Gomer, A. Ferrario, M. Luna, N. Rucker and S. Wong, Lasers Surg. Med., 2006, 38, 516–521 CrossRef PubMed.
- M. Zheng, C. Yue, Y. Ma, P. Gong, P. Zhao, C. Zheng, Z. Sheng, P. Zhang, Z. Wang and L. Cai, ACS Nano, 2013, 7, 2056–2067 CrossRef CAS PubMed.
- S. Fickweiler, R.-M. Szeimies, W. Bäumler, P. Steinbach, S. Karrer, A. E. Goetz, C. Abels and F. Hofstädter, J. Photochem. Photobiol., B, 1997, 38, 178–183 CrossRef CAS.
- X. Su, J. Fricke, D. G. Kavanagh and D. J. Irvine, Mol. Pharm., 2011, 8, 774–787 CrossRef CAS PubMed.
- A. S. Levine, M. Sivulich, P. H. Wiernik and H. B. Levy, Cancer Res., 1979, 39, 1645–1650 CAS.
- D. W. Siemann, Cancer Treat. Rev., 2011, 37, 63–74 CrossRef CAS PubMed.
- S. Koch, F. Mayer, F. Honecker, M. Schittenhelm and C. Bokemeyer, Br. J. Cancer, 2003, 89, 2133–2139 CrossRef CAS PubMed.
- J. K. Mohindra and A. M. Rauth, Cancer Res., 1976, 36, 930–936 CAS.
- Z. Wang and P. C. Ho, Small, 2010, 6, 2576–2583 CrossRef CAS PubMed.
- J. M. Chan, L. Zhang, R. Tong, D. Ghosh, W. Gao, G. Liao, K. P. Yuet, D. Gray, J.-W. Rhee and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2213–2218 CrossRef CAS PubMed.
- H. Tanjore and R. Kalluri, Am. J. Pathol., 2006, 168, 715 CrossRef CAS PubMed.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS PubMed.
- D. A. Nelson, T.-T. Tan, A. B. Rabson, D. Anderson, K. Degenhardt and E. White, Genes Dev., 2004, 18, 2095–2107 CrossRef CAS PubMed.
- D. Steinbach, S. Wittig, G. Cario, S. Viehmann, A. Mueller, B. Gruhn, R. Haefer, F. Zintl and A. Sauerbrey, Blood, 2003, 102, 4493–4498 CrossRef CAS PubMed.
- M. D. Norris, J. Smith, K. Tanabe, P. Tobin, C. Flemming, G. L. Scheffer, P. Wielinga, S. L. Cohn, W. B. London and G. M. Marshall, Mol. Cancer. Ther., 2005, 4, 547–553 CrossRef CAS PubMed.
- L. E. van Vlerken, Z. Duan, S. R. Little, M. V. Seiden and M. M. Amiji, AAPS J., 2010, 12, 171–180 CrossRef CAS PubMed.
- J. Jia, F. Zhu, X. Ma, Z. W. Cao, Y. X. Li and Y. Z. Chen, Nat. Rev. Drug Discovery, 2009, 8, 111–128 CrossRef CAS PubMed.
- P. Marino, A. Preatoni, A. Cantoni and G. Buccheri, Lung Cancer, 1995, 13, 1–12 CrossRef CAS.
- H. L. Wong, R. Bendayan, A. M. Rauth and X. Y. Wu, J. Controlled Release, 2006, 116, 275–284 CrossRef CAS PubMed.
- H. L. Wong, A. M. Rauth, R. Bendayan, J. L. Manias, M. Ramaswamy, Z. Liu, S. Z. Erhan and X. Y. Wu, Pharm. Res., 2006, 23, 1574–1585 CrossRef CAS PubMed.
- P. Zhang, G. Ling, X. Pan, J. Sun, T. Zhang, X. Pu, S. Yin and Z. He, Nanomedicine, 2012, 8, 185–193 CrossRef CAS PubMed.
- B. Weigelt, J. L. Peterse and L. J. Van't Veer, Nat. Rev. Cancer, 2005, 5, 591–602 CrossRef CAS PubMed.
- G. Poste and I. J. Fidler, Nature, 1980, 283, 139–146 CrossRef CAS.
- H. Clevers, Nat. Med., 2011, 313–319 CrossRef CAS PubMed.
- I. Brouet and H. Ohshima, Biochem. Biophys. Res. Commun., 1995, 206, 533–540 CrossRef CAS.
- L. I. Gold, Crit. Rev. Oncogenesis, 1998, 10, 303–360 Search PubMed.
- J. Park, S. H. Wrzesinski, E. Stern, M. Look, J. Criscione, R. Ragheb, S. M. Jay, S. L. Demento, A. Agawu, P. Licona Limon, A. F. Ferrandino, D. Gonzalez, A. Habermann, R. A. Flavell and T. M. Fahmy, Nat. Mater., 2012, 11, 895–905 CrossRef CAS PubMed.
- J. Gao, Y. Xia, H. Chen, Y. Yu, J. Song, W. Li, W. Qian, H. Wang, J. Dai and Y. Guo, Nanomedicine, 2014, 9, 279–293 CrossRef CAS PubMed.
- M. E. Werner, S. Karve, R. Sukumar, N. D. Cummings, J. A. Copp, R. C. Chen, T. Zhang and A. Z. Wang, Biomaterials, 2011, 32, 8548–8554 CrossRef CAS PubMed.
- J. V. Frangioni, J. Clin. Oncol., 2008, 26, 4012–4021 CrossRef PubMed.
- T. Islam and M. G. Harisinghani, Cancer Biomarkers, 2009, 5, 61–67 CAS.
- S. Aryal, J. Key, C. Stigliano, J. S. Ananta, M. Zhong and P. Decuzzi, Biomaterials, 2013, 34, 7725–7732 CrossRef CAS PubMed.
- Z. Liao, H. Wang, X. Wang, P. Zhao, S. Wang, W. Su and J. Chang, Adv. Funct. Mater., 2011, 21, 1179–1186 CrossRef CAS.
- A. J. Mieszawska, Y. Kim, A. Gianella, I. van Rooy, B. Priem, M. P. Labarre, C. Ozcan, D. P. Cormode, A. Petrov and R. Langer, Bioconjugate Chem., 2013, 24, 1429–1434 CrossRef CAS PubMed.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- J. A. Copp, R. H. Fang, B. T. Luk, C.-M. J. Hu, W. Gao, K. Zhang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13481–13486 CrossRef CAS PubMed.
- N. Kevin, Nanoscale, 2013, 5, 8884–8888 RSC.
- C.-M. J. Hu, S. Kaushal, H. S. T. Cao, S. Aryal, M. Sartor, S. Esener, M. Bouvet and L. Zhang, Mol. Pharm., 2010, 7, 914–920 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |