
Cu cluster-anchored bismuthene promoting electrocatalytic reduction of CO2 into C2 products: a theoretical study

Abstract
The electrocatalytic CO2 reduction reaction (eCO2RR) to value-added multicarbon (C2) represents a promising carbon-neutral pathway, yet designing efficient catalysts remains challenging. Although Cu-based materials are prominent for C2, their performance requires further optimization. Here, we employ density functional theory (DFT) to investigate atomically precise Cun clusters (n = 1–4) doped on bismuthene (monolayer Bi(001)) as tunable catalysts. Our computations reveal that Cu cluster doped Bi(001) significantly enhances the adsorption capability for key intermediates (COOH* and CO*) and significantly reduces the potential-limiting step (PLS) free energy for CHOCO* formation. However, for Cu1@Bi(001), the local coordination environment resembles that of pristine Bi(001), leading to a similar reaction mechanism 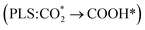 . As the size of Cu clusters increases (Cu2–Cu4), the active sites from Bi–Cu shift to Cu–Cu pairs, inducing a mechanistic shift to Cu(111)-like behavior (PLS: CO* → CHO*). Comparative PLS analysis reveals that Cu cluster doped systems outperform pristine Bi(001), and only controlled Cun clusters (n = 1–3) can effectively enhance the C2 selectivity of bismuthene, whereas excessive Cun cluster incorporation (n = 4) leads to suboptimal performance. Significantly, through energy and electronic structure analyses, we reveal that the adsorption energy differences of key intermediates, their electron transfer ratios and the “Cu–CHO*” bond strength serve as effective descriptors for PLS free energy, providing an indirect measure of catalytic performance. These findings establish bismuthene as a programmable platform for C2 synthesis, demonstrating how atomic-scale synergy between Cu clusters and 2D bismuthene substrates can overcome traditional scaling relations in eCO2RR catalysis.
. As the size of Cu clusters increases (Cu2–Cu4), the active sites from Bi–Cu shift to Cu–Cu pairs, inducing a mechanistic shift to Cu(111)-like behavior (PLS: CO* → CHO*). Comparative PLS analysis reveals that Cu cluster doped systems outperform pristine Bi(001), and only controlled Cun clusters (n = 1–3) can effectively enhance the C2 selectivity of bismuthene, whereas excessive Cun cluster incorporation (n = 4) leads to suboptimal performance. Significantly, through energy and electronic structure analyses, we reveal that the adsorption energy differences of key intermediates, their electron transfer ratios and the “Cu–CHO*” bond strength serve as effective descriptors for PLS free energy, providing an indirect measure of catalytic performance. These findings establish bismuthene as a programmable platform for C2 synthesis, demonstrating how atomic-scale synergy between Cu clusters and 2D bismuthene substrates can overcome traditional scaling relations in eCO2RR catalysis.

 Please wait while we load your content...
Please wait while we load your content...

