
Correlating the structural transformation and properties of ZIF-67 during pyrolysis, towards electrocatalytic oxygen evolution†

Abstract
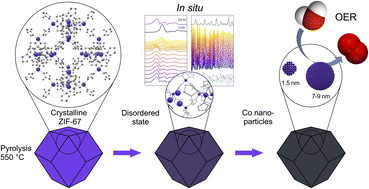
There is an emerging interest in using pyrolyzed metal–organic frameworks (MOFs) for electrocatalytic applications. While the MOF precursor and the final pyrolyzed catalyst are usually investigated, the pyrolysis process itself is often treated as a ‘black box’, and the phase transition is poorly understood as a result. The process further depends on the specific experimental setup, in terms of e.g. the heating ramp, carrier gas and heat- and mass transport, complicating the comparison of catalyst properties across the literature. In this study, we use in situ X-ray absorption spectroscopy and total scattering to elucidate the thermal decomposition of ZIF-67 to a MOF-derived nanomaterial (MDN), which is composed of cobalt nanoparticles embedded in a carbonaceous matrix. Furthermore, we demonstrate that the phase transition can be halted at different stages, allowing the corroboration of the in situ analyses with properties of ex situ MDNs produced by pyrolyzing ZIF-67 in a tube furnace at various temperatures and durations. The electrochemical properties of the ex situ MDNs were studied systematically towards the oxygen evolution reaction (OER), facilitating the correlation of catalyst properties and structural characteristics for the ZIF-67 based catalysts. Although ZIF-67 is generally acclaimed for its thermal stability up to approx. 400 °C, this study demonstrates the emergence of disorder at lower temperatures (approx. 150 °C). This disorder manifests as distortions of the framework by contraction of nearest neighbor Co–N bond distance, and an initial formation of cobalt clusters. Although this disorder resulted in poorer electrocatalytic performance relative to pristine ZIF-67, extending the temperature and duration of the pyrolysis process produced MDNs with superior electrochemical properties relative to pristine ZIF-67. The best catalyst exhibits the lowest overpotential against the oxygen evolution reaction of 416 mV, which is an improvement of 200 mV compared to the pristine MOF catalyst. The more heavily pyrolyzed samples are composed of cobalt nanoparticles with a bimodal size distribution at 1.4 nm and 7.1 nm (pyrolyzed at 550 °C for 8 or 12 h) and 1.7 nm and 9.4 nm (pyrolyzed at 650 °C) confined in a carbonaceous matrix and with BET surface areas of 40–124 m2 g−1.
- This article is part of the themed collections: Functional Framework Materials, 2024 Journal of Materials Chemistry A Most Popular Articles and Celebrating International Women’s day 2024: Women in Materials Science
 Please wait while we load your content...
Please wait while we load your content...

