
Theoretical investigation of the non-metal sites of two-dimensional conjugated metal–organic frameworks based on benzenehexathiol for hydrogen evolution activity enhancement†

Abstract
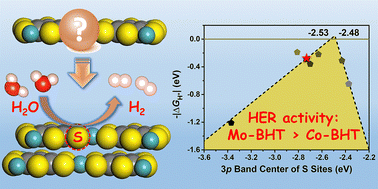
For electrocatalysts, the electrocatalytic activity of the non-metal sites is not negligible. We found that sulfur atoms should be the predominant active site for conjugated metal–organic frameworks (c-MOFs) based on benzenehexathiol (BHT) toward the hydrogen evolution reaction (HER). There is a “volcano”-shaped relationship between their HER activity and 3p band center of the sulfur active site. Interlayer interactions are also crucial in determining the HER activity of c-MOFs. Based on these findings, we proposed that Mo–BHT possesses excellent potential as an active HER catalyst.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry C HOT Papers and Celebrating ten years of Journal of Materials Chemistry C
 Please wait while we load your content...
Please wait while we load your content...

