
Thermally stable and rewritable circularly polarized luminescent helical poly(diphenylacetylene)s: stabilization of macromolecular helicity memory via reversible ion-pair formation†
 *bc
*bc
Abstract
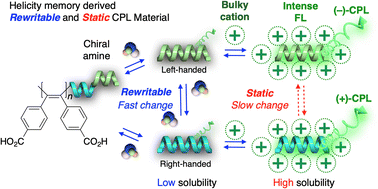
Fluorescent poly(diphenylacetylene) (poly-1-H) bearing carboxy pendants forms a one-handed helical conformation upon thermal annealing in the presence of nonracemic chiral amines in water. This macromolecular helicity is retained (memorized) even after complete removal of the amines (h-poly-1-H); however, it is easily lost at high temperatures. In this study, we report an efficient stabilization of the helicity memory of h-poly-1-H even at high temperatures together with a significant enhancement in the solubility and fluorescent properties of h-poly-1-H using the reversible formation of an ion-pair complex with bulky ammonium or phosphonium cations. The ion-paired polymer with tetradecylammonium cations (h-poly-1-A10) nearly retained its helicity memory even after heating at 80 °C for 24 h or at 100 °C for 2 h in toluene, and significant enhancement in fluorescence quantum yield from 9% to 32% was observed. In addition, h-poly-1-A10 formed a tough self-supporting film exhibiting strong CPL emission because of its helicity memory, and its ethanol solution could be used as a CPL ink. Because the stabilization of the helicity memory by noncovalent ion-pair formation is reversible, the ion-paired polymer could be reconverted to the original carboxylic-acid-type polymer while maintaining the helicity memory. Moreover, its macromolecular helicity can be repeatedly switched through heat treatment in the presence of the enantiomeric counterpart of the chiral amines and then further stabilized by the ion-pair formation.
- This article is part of the themed collection: Circularly Polarised Luminescence
 Please wait while we load your content...
Please wait while we load your content...

