
Sublimable complexes with spin switching: chemical design, processing as thin films and integration in graphene-based devices†
 ‡a
Francisco Javier
Valverde-Muñoz,
‡a
Rosa
Córdoba,
a
M. Carmen
Muñoz,
b
Javier
Herrero-Martín,
c
José Antonio
Real
*a
and
Eugenio
Coronado
*a
‡a
Francisco Javier
Valverde-Muñoz,
‡a
Rosa
Córdoba,
a
M. Carmen
Muñoz,
b
Javier
Herrero-Martín,
c
José Antonio
Real
*a
and
Eugenio
Coronado
*a
Abstract
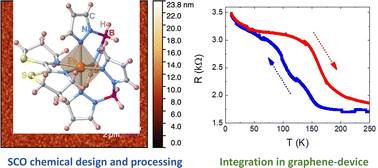
Among the different types of switchable molecular compounds, sublimable Fe(II) SCO molecules provide a suitable platform to develop smart devices that respond to external stimuli. Herein, we report the synthesis, crystallographic structure and magnetic properties of three new neutral Fe(II) SCO molecules belonging to the {Fe[H2B(pz)2]2(L)} family with bidentate-α-diimine ligands L = 3-(pyridin-2-yl)-[1,2,3]triazolo[1,5-a]pyridine (tzpy), 5,5′,6,6′-tetrahydro-4H,4′H-2,2′-bi(1,3-thiazine) (btz) and 4,4′,5,5′-tetrahydro-2,2′-bithiazole (bt) (1, 2 and 3, respectively), as well as two solvated forms of 1 and 3. All three desolvated compounds present thermal- and light-induced SCO transitions with different degrees of cooperativity and effectiveness. Furthermore, 1 and 2 are demonstrated to be sublimable under HV conditions, affording homogeneous thin films 200 nm thick (TF1 and TF2) that retain the chemical integrity of the original molecules regardless of the deposition surface. The SCO behaviour of the films is characterized by the XAS technique revealing the partial retainment of both thermal- and light-induced spin transitions, yet losing the cooperativity. Finally, SCO/2D horizontal hybrid devices based on CVD-graphene are produced using these films. Being the first ones of this type utilizing molecules of the {Fe[H2B(pz)2]2(L)} family, with L = tzpy and btz, the devices have allowed the successful detection of the thermal SCO transition through the electric properties of CVD-graphene.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry C Most Popular Articles and Celebrating ten years of Journal of Materials Chemistry C
 Please wait while we load your content...
Please wait while we load your content...

