
Hydrogen-bonded organic framework-stabilized charge transfer cocrystals for NIR-II photothermal cancer therapy†

Abstract
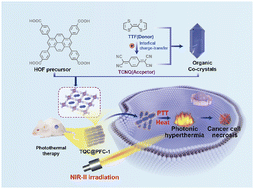
Charge-transfer (CT) cocrystals consisting of an electron donor and acceptor have gained attention for designing photothermal (PT) conversion materials with potential for biomedical and therapeutic use. However, the applicability of CT cocrystals is limited by their low stability and aqueous dispersity in biological settings. In this study, we present the self-assembly of CT cocrystals within hydrogen-bonded organic frameworks (HOFs), which not only allows for the dispersion and stabilization of cocrystals in aqueous solution but also promotes the CT interaction within the confined space of HOFs for photothermal conversion. We demonstrate that the CT interaction-driven self-assembly of tetrathiafulvalene (TTF) and tetracyanoquinodimethane (TCNQ) with PFC-1 HOFs results in the formation of cocrystal-encapsulated TQC@PFC-1 while retaining the crystalline structure of the cocrystal and PFC-1. TQC@PFC-1, in particular, exhibits significant absorption in the second near-infrared region (NIR-II) and excellent photothermal conversion efficiency, as high as 32%. Cellular delivery studies show that TQC@PFC-1 can be internalized in different types of cancer cells, leading to an effective NIR-II photothermal therapy effect both in cultured cells and in vivo. We anticipate that the strategy of self-assembly and stabilization of CT cocrystals in nanoscale HOFs opens the path for tuning their photophysical properties and interfacing cocrystals with biological settings for photothermal therapeutic applications.
- This article is part of the themed collections: Functional Framework Materials for Biomedical Applications and #MyFirstJMCB
 Please wait while we load your content...
Please wait while we load your content...

