
Highly modular hepatitis B virus-like nanocarriers for therapeutic protein encapsulation and targeted delivery to triple negative breast cancer cells†
 a
Wilfred
Chen
*a
a
Wilfred
Chen
*a
Abstract
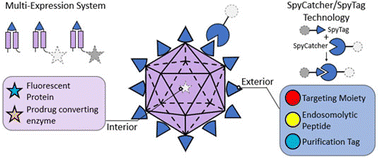
Protein therapeutics offer enormous clinical impact in treating a variety of diseases by offering high selectivity with limited off-target effects. However, delivery challenges severely hinder functional proteins from reaching their target cells and necessitate frequent administration. To address these problems, nanocarrier encapsulation can provide protease protection and enhanced targeted transportation of functional proteins to their intended disease site. Inspired by their viral analogues, virus-like particles (VLPs) are non-infectious viral capsids that have potential for drug delivery applications because of their shared structural characteristics, such as high loading capacity, particle stability, and structural uniformity. Here, we describe a modular hepatitis B virus (HBV) VLP delivery platform offering tunable modifications of both the exterior and interior viral capsid surfaces via SpyCatcher–SpyTag bioconjugation and a multi-expression system, respectively. This new platform facilitates modification with epidermal growth factor receptor (EGFR)-targeting proteins and encapsulation with both model green fluorescent protein (GFP) and prodrug-converting yeast cytosine deaminase (yCD) enzyme. The resultant targeted VLPs demonstrated enhanced uptake and toxicity in EGFR-overexpressing triple negative breast cancer (TNBC) cells in contrast to non-malignant breast epithelial cells.
- This article is part of the themed collections: Materials, Physical and Biological Chemistry of Protein Cages, 2023 Journal of Materials Chemistry B HOT Papers and #MyFirstJMCB
 Please wait while we load your content...
Please wait while we load your content...

