
Tuning properties of biocatalysis using protein cage architectures
 a
and
Trevor
Douglas
*a
a
and
Trevor
Douglas
*a
Abstract
Compartmentalization of cellular activities is an extremely important mechanism within cells, across all domains of life, for high efficiency of cell function. Bacterial microcompartments are exemplary protein-based cage structures that act as subcellular compartments encapsulating biocatalysts. They are able to achieve segregation of metabolic reactions from the bulk environment, which can alter the properties (including efficiency and selectivity) of biochemical processes and enhance overall cell function. By mimicking these naturally occurring compartments using protein cage platforms, synthetic catalytic materials have been made to achieve well-defined biochemical catalysis with desired and enhanced activities. This Perspective reviews the study in the past decade or so on artificial nanoreactors developed based on protein cage architectures, and summarizes the effects of protein cages on the properties of encapsulated enzymatic catalysis, including reaction efficiency and substrate selectivity. Given the significance of metabolic pathways in living systems and its inspiration in biocatalysis, our perspectives are also presented on cascade reactions, which are illustrated from three aspects: the technical challenges of controlling molecular diffusion to achieve the desired properties of multistep biocatalysis, the solutions to these challenges presented by nature, and how biomimetic approaches have been adopted in the design of biocatalytic materials using protein cage architectures.
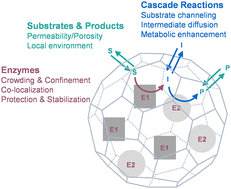
- This article is part of the themed collections: Materials, Physical and Biological Chemistry of Protein Cages, 2023 Journal of Materials Chemistry B HOT Papers and Biocatalysis: A cross-journal collection
 Please wait while we load your content...
Please wait while we load your content...

