
Cell-binding peptides on the material surface guide stem cell fate of adhesion, proliferation and differentiation†

Abstract
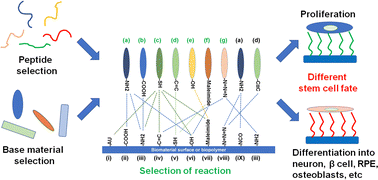
Human cells, especially stem cells, need to communicate and interact with extracellular matrix (ECM) proteins, which not only serve as structural components but also guide and support cell fate and properties such as cell adhesion, proliferation, survival and differentiation. The binding of the cells with ECM proteins or ECM-derived peptides via cell adhesion receptors such as integrins activates several signaling pathways that determine the cell fate, morphological change, proliferation and differentiation. The development of synthetic ECM protein-derived peptides that mimic the biological and biochemical functions of natural ECM proteins will benefit academic and clinical application. Peptides derived from or inspired by specific ECM proteins can act as agonists of each ECM protein receptor. Given that most ECM proteins function in cell adhesion via integrin receptors, many peptides have been developed that bind to specific integrin receptors. In this review, we discuss the peptide sequence, immobilization design, reaction method, and functions of several ECM protein-derived peptides. Various peptide sequences derived from mainly ECM proteins, which are used for coating or grafting on dishes, scaffolds, hydrogels, implants or nanofibers, have been developed to improve the adhesion, proliferation or differentiation of stem cells and to culture differentiated cells. This review article will help to inform the optimal choice of ECM protein-derived peptides for the development of scaffolds, implants, hydrogels, nanofibers and 2D cell culture dishes to regulate the proliferation and direct the differentiation of stem cells into specific lineages.
- This article is part of the themed collections: Journal of Materials Chemistry B Recent Review Articles and 2024 Journal of Materials Chemistry B Lunar New Year
 Please wait while we load your content...
Please wait while we load your content...

