
Fe(iii)-carboxythiolate layered metal–organic frameworks with interest as active materials for rechargeable alkali-ion batteries†
 ‡a
Long H. B.
Nguyen,
bd
Nathalie
Guillou,
c
Nicolas
Dupré,
a
Christophe
Payen,
a
Nicolas
Louvain,
bd
Lorenzo
Stievano,
bd
Philippe
Poizot
a
and
Thomas
Devic
*a
‡a
Long H. B.
Nguyen,
bd
Nathalie
Guillou,
c
Nicolas
Dupré,
a
Christophe
Payen,
a
Nicolas
Louvain,
bd
Lorenzo
Stievano,
bd
Philippe
Poizot
a
and
Thomas
Devic
*a
Abstract
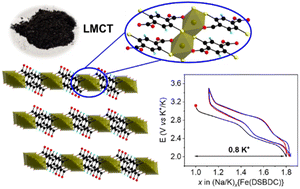
Metal organic frameworks (MOFs) built up from metal–sulfur (M–S) bonds have shown great promise in the last decade thanks to their impressive electronic properties arising from the more covalent nature of the M–S bond when compared to M–O. In this study, we aim to expand the scope of available S-based MOFs to high oxidation state cations Mn+ (n ≥ 3). Through the systematic exploration of the reactivity of the mixed thiolate-carboxylate ligand 2,5-disulfhydrylbenzene-1,4-dicarboxylic acid (H4DSBDC) with Fe(III) salts, three new compounds were isolated. The structure of these materials consists of the same hybrid layers made of chains of edge-sharing FeS4O2 octahedra connected by fully deprotonated DSBDC4−, alternating with either organic (dimethyammonium, H2N(CH3)2+) or inorganic (Na+ or K+) cations. The Fe–S connection leads to a strong absorption in the visible range, while the +III oxidation state of Fe, evidenced by Mössbauer spectroscopy, ensures a fairly good stability towards oxygen and water, higher than that typically observed with +II cations. Furthermore, Na{Fe(DSBDC)} exhibits an interesting reversible electrochemical activity when cycled both versus Li, Na and K in half-cells with good capacity retention, confirming the potential of this family of materials for applications involving electron transfer.
- This article is part of the themed collection: Functional Framework Materials
 Please wait while we load your content...
Please wait while we load your content...

