
Metal–organic framework-derived CuO catalysts for the efficient hydrogenolysis of hardwood lignin into phenolic monomers†
 *a
Run-Cang
Sun
*a
*a
Run-Cang
Sun
*a
Abstract
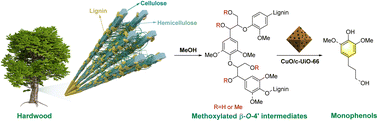
The selective reductive catalytic deconstruction (RCD) of lignin into phenolic monomers provides the possibility for making the full use of lignocellulose. However, the widespread use of precious metal catalysts and the harsh reaction conditions present the challenge of poor industrial utilization in the current research. Herein, we report a metal–organic framework (MOF)-derived copper oxide catalyst (CuO/c-UiO-66), which exhibits superior catalytic properties in the RCD of hardwood lignin and affords high yields (up to 42.8 wt%) of monomeric phenols via the C–O bond scission. The mechanistic reactions using lignin model compounds reveal that phenolic compounds with propyl or propanol end chains are selectively produced during the catalytic hydrogenolysis reaction. The enhanced catalytic reactivity is attributed to the synergy of acid and base sites of the catalyst, which facilitates the C–O bond cleavage process. The new insights of this study provide guidance toward the rational design of Cu-based catalysts for RCD of lignin.
- This article is part of the themed collections: Functional Framework Materials and 2023 Journal of Materials Chemistry A HOT Papers
 Please wait while we load your content...
Please wait while we load your content...

