
Long-term cycling stability of a SnS2-based covalent organic nanosheet anode for lithium-ion batteries†

Abstract
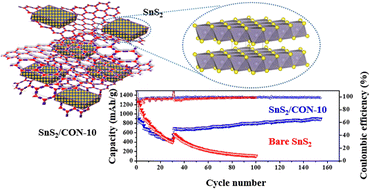
Various SnS2-based carbonaceous anodes for lithium ion battery (LIB) systems have been developed to enhance the electrochemical performance of SnS2 materials and to overcome the disadvantages of transition metal sulfides with less interfacial surface sites and low electrochemical conductivity. In this study, we introduced a new strategy of hybridization of SnS2 and covalent organic nanosheets (CONs) that have high flexibility, high stability in organic electrolytes, and many interfacial surface sites. The CON provided reaction sites for the growth of SnS2 nanoparticles due to the strong electrostatic interaction between the sulfur heteroatoms of CONs and Sn4+, resulting in the formation of ultrathin SnS2 nanoplates on the CON nanosheets. The resulting SnS2-based CON showed outstanding cyclic stability over 5600 charge/discharge cycles at a current density of 1.0 A g−1 in the LIB system. In particular, the prominent interfacial surface sites of CONs provided large accessible areas for lithium ions, showing stable successive cycling performances with improved electrical and ionic conductivities.
- This article is part of the themed collection: 1D/2D materials for energy, medicine, and devices
 Please wait while we load your content...
Please wait while we load your content...

