
Fusing a machine learning strategy with density functional theory to hasten the discovery of 2D MXene-based catalysts for hydrogen generation†
 ‡a
Jayant K.
Singh
*ab
‡a
Jayant K.
Singh
*ab
Abstract
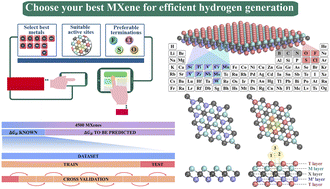
The complexity of the topological and combinatorial configuration space of MXenes can give rise to gigantic design challenges that cannot be addressed through traditional experimental or routine theoretical methods. To this end, we establish a robust and more broadly applicable multistep workflow using supervised machine learning (ML) algorithms to construct well-trained data-driven models for predicting the hydrogen evolution reaction (HER) activity of 4500 MM′XT2-type MXenes, where 25% of the materials space (1125 systems) is randomly selected to evaluate the HER performance using density functional theory (DFT) calculations. As the most desirable ML model, the gradient boosting regressor (GBR) processed with recursive feature elimination (RFE), hyperparameter optimization (HO) and the leave-one-out (LOO) approach accurately and rapidly predicts the Gibbs free energy of hydrogen adsorption (ΔGH) with a low predictive mean absolute error (MAE) of 0.358 eV. Based on these observations, the H atoms adsorbed directly on top of the outermost metal-atom layer of the MM′XT2-type MXenes (site 1) with Nb, Mo and Cr metals with O functionalization are discovered to be highly stable and active for catalysis, surpassing commercially available platinum-based counterparts. Overall, the physically meaningful predictions and insights of the developed ML/DFT-based multistep workflow will open new avenues for accelerated screening, rational design and discovery of potential HER catalysts.
- This article is part of the themed collections: Machine Learning and Artificial Intelligence: A cross-journal collection, 2023 Journal of Materials Chemistry A Most Popular Articles and #MyFirstJMCA
 Please wait while we load your content...
Please wait while we load your content...

