
One-dimensional III-nitrides: towards ultrahigh efficiency, ultrahigh stability artificial photosynthesis
 *a
and
*a
and
Abstract
The depletion of carbon-based fuels and emerging environmental problems are more than ever driving the demand for green energy sources. Artificial photosynthesis demonstrated by catalytically active semiconductors can convert solar energy into storable energy sources such as hydrogen and carbon products by water splitting and carbon dioxide reduction reaction, respectively. Semiconductors with one-dimensional (1D) nanostructures have gained tremendous attention due to their large surface area and enhanced light absorption. However, semiconductor nanostructures suffer from degradation caused by oxidation, corrosion, and undesirable changes in morphology during reaction in aqueous electrolytes. In this review, 1D nanostructured III-nitrides are introduced to offer a comprehensive overview of their synthesis methods, unique structural, electronic, optical, surface, and catalytic properties, as well as their efficiency, and stability for artificial photosynthesis. Band gap engineering and tandem stacking of 1D III-nitrides for broadband light absorption are highlighted. Recent findings on the inherent catalytic activity and stability of 1D III-nitrides are presented. Furthermore, this paper summarizes the ability of cocatalysts integrated on 1D III-nitrides to overcome the performance bottlenecks of water splitting and CO2 reduction reactions. Finally, the challenges of 1D III-nitrides for practical application of photo(electro)chemical reactions and prospective insights for future development are provided.
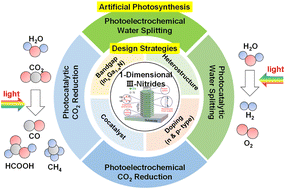
- This article is part of the themed collections: 1D/2D materials for energy, medicine, and devices and Journal of Materials Chemistry A Recent Review Articles
 Please wait while we load your content...
Please wait while we load your content...

