
Fe, Cu dual-metal single atom catalyst on commercial carbon black for efficient oxygen reduction reaction†

Abstract
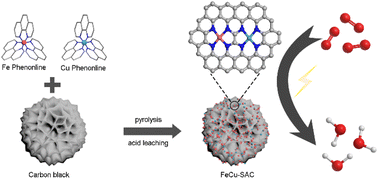
3d transition metal single atom catalysts (SACs) (e.g. M = Fe, Co, Cu) are considered as promising catalysts for oxygen reduction reaction (ORR), among which Fe SACs display comparable performance to commercial Pt/C. However, Fe SACs still demand significant improvements in activity and stability for further industrial deployment. Herein, a Fe, Cu dual metal SAC was synthesized using a facile ligand-mediated method. Fe and Cu atoms were atomically dispersed on commercial carbon black in the form of FeN4 and CuN4 sites. Owing to the interactions between the adjacent Fe and Cu atoms co-existing in a very tiny area, the as-synthesized Fe, Cu dual metal SAC exhibited excellent ORR performance with a high half-wave potential of 0.926 V and good stability in 0.1 M KOH, which are significantly higher than that of the Fe-SAC or Cu-SAC control samples. Density function theory calculations showed that the Cu single atom can act as an electron donator enriching the electron density of Fe sites, thus endowing the Fe sites with optimized adsorption/desorption energy for ORR intermediates and thereby facilitating the ORR activity of FeCu-SAC. Moreover, a study on three typical commercial carbon substrates indicated that carbon support with high surface areas and hierarchical pore structure was beneficial for ORR. The excellent ORR activity and durability of FeCu-SAC enabled the constructed Zn–air battery to have a high power density of 201.4 mW cm−2 and high specific energy of 827.68 W h kg−1.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry A Most Popular Articles and 2024 Journal of Materials Chemistry A Lunar New Year collection
 Please wait while we load your content...
Please wait while we load your content...

