
A highly permeable porous organic cage composite membrane for gas separation†

Abstract
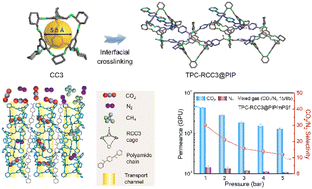
Membrane-based separation processes have become the focus of research in the field of CO2/N2 and CH4/N2 separation due to their green, energy-saving, and high-efficiency characteristics. However, the insufficient gas permeance of separation membranes greatly restricts their applications in practical industrial separation. In this work, a highly permeable porous organic cage (POC) composite membrane was first proposed and constructed with the RCC3 porous organic cage crosslinked by terephthaloyl chloride (TPC). The RCC3 porous organic cage displayed a pore size of approximately 5.4 Å and a high specific surface area of 442.3 m2 g−1, which provided amine-rich subnanochannels for the rapid penetration of CO2. Moreover, the interfacial crosslinking reaction between RCC3 and TPC enabled the assembly of the TPC-RCC3 ultrathin film on the surface of the modified polysulfone (mPSf) substrate. On this basis, a trace amount of piperazine anhydrous (PIP) was further employed to regulate the cross-linking degree of the ultrathin film for improved CO2/N2 selectivity. The as-prepared composite membrane displayed a high CO2 permeance of 4303 GPU with a CO2/N2 selectivity of 30, and CH4 permeance of 1216 GPU with a CH4/N2 selectivity of 3.0 at 1 bar, and could maintain the permselectivity under a long-term operation. The excellent separation performance provided a more economical solution for CO2 capture from flue gas or natural gas purification.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry A Most Popular Articles and 2024 Journal of Materials Chemistry A Lunar New Year collection
 Please wait while we load your content...
Please wait while we load your content...

