
Modulating oxygen vacancy concentration on Bi4V2O11 nanorods for synergistic photo-driven plastic waste oxidation and CO2 reduction†
 *b
*b
Abstract
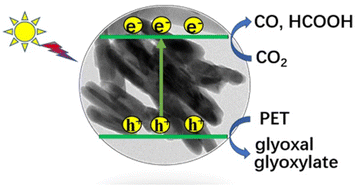
Sunlight-driven CO2 reduction coupled with photo-oxidation of plastic waste into value-added chemicals is a very attractive approach towards solving the greenhouse and environmental crisis. Herein, Bi4V2O11 nanorods with regulable O-vacancy concentration have been synthesized by the solvothermal method, aiming to provide abundant active sites for CO2 adsorption and boost the separation of photogenerated carriers. In a dual-function system, gas production (CO) mainly from the CO2 reduction-half-reaction reaches 64.7 μmol g−1 h−1 on Bi4V2O11 with rich oxygen vacancies (VO-BVO-15) in PET hydrolysis solution under 300 W Xe lamp irradiation, 24.5-fold higher than that in 2 M KOH solution. Moreover, a considerable amount of HCOOH product with a conversion rate of 0.7 mmol gcata.−1 is also achieved under 5 h of irradiation. Glyoxal (6.9 mmol gcata.−1) and glyoxylate (3.2 mmol gcata.−1) are produced mainly from the PET oxidation-half-reaction. This work presents an in-depth study of the development of Bi–O–V photocatalysts through defect engineering for photocatalytic CO2 reduction and demonstrates a promising strategy for reuse of plastic waste and realizing carbon cycle with low energy consumption.
- This article is part of the themed collections: Polymer Upcycling and Celebrating ten years of Journal of Materials Chemistry A
 Please wait while we load your content...
Please wait while we load your content...

