
Encapsulated C12A7 electride material enables a multistep electron transfer process for cross-coupling reactions†

Abstract
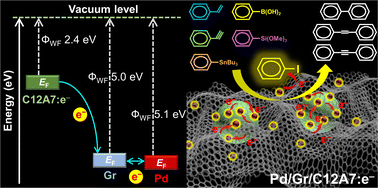
The electronic structures of active sites fundamentally determine the catalytic performance in chemical reactions and are also crucial for obtaining a detailed understanding of charge transport and reaction mechanisms. In this study, the regulation of the electronic structure of active metal Pd can be achieved through a multi-step electron transfer process formed by a synergy of [Ca24Al28O64]4+(e−)4 (C12A7:e−) electride and conductive graphene (Gr). The composite catalytic system (Pd/Gr/C12A7:e−) significantly facilitates the transfer of electrons from electron-rich Pd active sites to aryl halides in Suzuki-coupling reactions, which enables superior catalytic performance with TOFs above 20 times higher than well-studied negatively charged Pd catalysts. No catalytic degradation was observed even after impregnating the catalyst in water because of the well-protected C12A7:e− electride by Gr. The present efficient catalyst can further trigger various carbon–carbon cross-coupling reactions with high activities. These results provide significant advantages for expanding the potential applications of electride materials, thereby allowing precise control of the electronic structure of the active sites and aiding in tuning the reaction conditions using a simple method.
- This article is part of the themed collection: Celebrating ten years of Journal of Materials Chemistry A
 Please wait while we load your content...
Please wait while we load your content...

