
Few-layer MoS2 nanosheets with and without silicon nanoparticles as anodes for lithium-ion batteries†
 *ab
Mike
Tebyetekerwa,
c
Nunzio
Motta,
bd
Jose A.
Alarco,
bd
Zhen-Jiang
He,
e
Jun-Chao
Zheng,
e
Aijun
Du
bd
and
Cheng
Yan
*ab
*ab
Mike
Tebyetekerwa,
c
Nunzio
Motta,
bd
Jose A.
Alarco,
bd
Zhen-Jiang
He,
e
Jun-Chao
Zheng,
e
Aijun
Du
bd
and
Cheng
Yan
*ab
Abstract
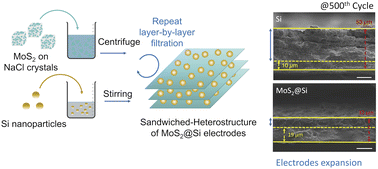
Few-layer two-dimensional (2D) molybdenum disulfide (MoS2) nanosheets are potential anode materials for lithium-ion batteries due to their stable electrochemical performance. On the other hand, silicon (Si) is attracting the attention of battery research due to its high specific capacity, but its problem of volume expansion remains a challenge. Hence, rationally designed MoS2 nanosheets with Si may take the synergy between these materials, which can not only mitigate the volume expansion but also maintain excellent electrochemical properties. Therefore, we have successfully synthesized few-layer MoS2 nanosheets on water-soluble, naturally abundant, and cost-effective recrystallized three-dimensional sodium chloride (NaCl) crystals. The obtained free-standing few-layer MoS2 nanosheets are utilized to form a MoS2@Si heterostructure. It has been confirmed that the layered MoS2 nanosheets can accommodate the volume expansion of Si and provide channels for lithium-ion transport during electrochemical cycles. After 500 cycles, the volume expansion is reduced to 68%, a remarkable improvement compared to 430% in pristine Si. Exceptional rate and cycling performance have been achieved in the composite anode, with a capacity retention of 60% compared to 0.3% for pristine Si at a high current density of 500 mA g−1. This work provides a rational design for Si and 2D material composites as anode materials for improved electrochemical performance.
- This article is part of the themed collections: 1D/2D materials for energy, medicine, and devices and 2023 Journal of Materials Chemistry A Most Popular Articles
 Please wait while we load your content...
Please wait while we load your content...

