
Tailoring stability, catalytic activity and selectivity of covalent metal–organic frameworks via steric modification of metal nodes†
 *a
and
Dan
Li
*a
*a
and
Dan
Li
*a
Abstract
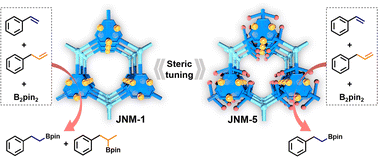
Although many efforts have been made to tune catalytic performance via the modification of MOF nodes including metal exchange, defect creation and metal insertion, steric tuning of MOF nodes via ligand modification remains challenging and unexplored. Herein, we have successfully fabricated two two-dimensional (2D) Cu(I) cyclic trinuclear unit (Cu-CTU)-based MOFs with similar structures, denoted as JNM-1 and JNM-5. JNM-1 has less steric hindrance on copper open sites, while JNM-5 incorporates bulky groups enhancing steric hindrance and partial coverage on copper open sites. Due to the steric effect, JNM-5 exhibited much higher crystallinity, porosity and chemical stability, but lower catalytic activity for hydroboration reactions than JNM-1. Interestingly, JNM-5 delivered much higher substrate selectivity and chemo-selectivity for hydroboration of olefins compared to JNM-1. Owing to its high chemical stability, JNM-5 can be reused for at least five cycles without losing catalytic performance and crystallinity, while the catalytic activity of JNM-1 is greatly decreased and it turns into an amorphous material after five cycles.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry Lectureship shortlisted candidates and Celebrating ten years of Journal of Materials Chemistry A
 Please wait while we load your content...
Please wait while we load your content...

