
Improving the intrinsic conductivity of δ-MnO2 by indium doping for high-performance neutral aqueous sodium-ion supercapacitors with commercial-level mass-loading†
 d
and
Junjie
He
*ac
d
and
Junjie
He
*ac
Abstract
Due to their potential use in energy storage and next-generation water purification devices, neutral aqueous supercapacitors (NASCs) have attracted significant attention from researchers. δ-MnO2 is a well-known cathode material for supercapacitors due to its 2D structure and high capacitance. However, its poor conductivity limits the mass loading on the electrode. Moreover, the dissolution of δ-MnO2 hampers its cycling stability and applicability in water purification. Here, indium is introduced to δ-MnO2 with the aid of iron. The crystal water and conductivity of the δ-MnO2 are significantly influenced by indium. After optimization of the concentration of indium in δ-MnO2, the gravimetric capacitance of δ-MnO2 increases to 1302 F g−1 (95% of the theoretical limit). At a mass loading of 10.6 mg cm−2, 180 F g−1, 1.9 F cm−2 and 54 F cm−3 at 10 mA cm−2 can be obtained. A NASC (In doped δ-MnO2//active carbon) exhibits a maximum areal energy density of 0.55 mW h cm−2 and a volumetric energy density of 18.7 mW h cm−3 at an areal power density of 1.0 mW cm−2. 90% capacitance retention after 13 000 cycles arises from the prevention of Jahn–Teller distortion. First principles calculations and experimental results demonstrate that In doping is effective in enhancing the intrinsic properties of δ-MnO2. These findings open up a new path towards high-performance supercapacitors.
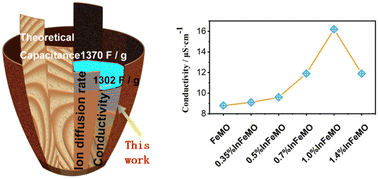
- This article is part of the themed collections: 1D/2D materials for energy, medicine, and devices and 2023 Journal of Materials Chemistry A HOT Papers
 Please wait while we load your content...
Please wait while we load your content...

