
Synthesis of platinum nanoparticles on strontium titanate nanocuboids via surface organometallic grafting for the catalytic hydrogenolysis of plastic waste†
 a
Robert M.
Kennedy,
ab
Xun
Wu,
cd
Frédéric A.
Perras,
d
A. Jeremy
Kropf,
a
Jens
Niklas,
a
Oleg G.
Poluektov,
a
Jianguo
Wen,
*f
Wenyu
Huang,
*cd
Aaron D.
Sadow,
*cd
Massimiliano
Delferro
*ag
and
a
Robert M.
Kennedy,
ab
Xun
Wu,
cd
Frédéric A.
Perras,
d
A. Jeremy
Kropf,
a
Jens
Niklas,
a
Oleg G.
Poluektov,
a
Jianguo
Wen,
*f
Wenyu
Huang,
*cd
Aaron D.
Sadow,
*cd
Massimiliano
Delferro
*ag
and
Abstract
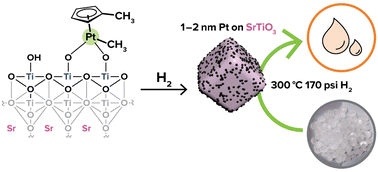
Pt/SrTiO3 (Pt/STO), prepared on small scale by atomic layer deposition (ALD), is a capable heterogeneous catalyst for the selective hydrogenolysis of polyolefins to hydrocarbon oils, providing a promising approach for upcycling plastic waste. However, because deposition by ALD is costly and resource-intensive, a new synthesis of Pt/STO is needed to effectively scale catalyst production and pursue the commercialization of upcycling processes. To that effect, this work details a scalable deposition method for Pt/STO made by surface organometallic chemistry (SOMC) techniques using Pt(II) acetylacetonate or and trimethyl(methylcyclopentadienyl)platinum. The STO support was calcined (550 °C), treated with ozone (200 °C), and finally steamed (200 °C) to afford a clean STO surface populated with only hydroxyl groups. Pt precursors were dissolved in toluene and deposited onto STO. After reduction at 300 °C, the STO support was decorated with 1.0–1.5 nm Pt nanoparticles. The size and loading of these nanoparticles were varied by employing a multi-cycle deposition and oxidation and/or reduction process designed to ALD techniques. These Pt/STO catalysts hydrogenolyzed isotactic polypropylene into liquid products (>95% yield) with average molecular weights of 200–300 Da (∼25 carbon atoms) and narrow size distributions at 300 °C and 180 psi H2.
- This article is part of the themed collections: Polymer Upcycling and 2023 Journal of Materials Chemistry A Most Popular Articles
 Please wait while we load your content...
Please wait while we load your content...

