
An unusual trans-hydrosilylation of prochiral 1,1-disubstituted cyclopropenes revealing the different nature of asymmetric palladium and rhodium catalysis†
 *ab
*ab
Abstract
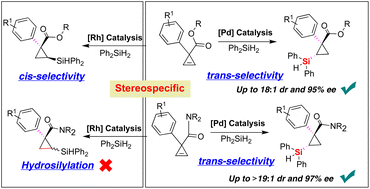
Catalytic asymmetric hydrosilylation is an extremely important and atom-economical chemical transformation in the field of catalysis and synthetic chemistry. Although stereospecific hydrosilylation provides a general strategy for elaborating diastereo- and enantioselective synthesis of optically pure organosilicon compounds, existing methods for accessing Si–C bond-forming hydrosilylation of alkenes rely almost entirely on terminal olefins. Herein, we reported a highly enantioselective palladium-catalyzed hydrosilylation reaction of 1,1-disubstituted carbonyl cyclopropenes with dihydrophenylsilane. We demonstrated that the palladium catalyst system provided stereodivergence to enable the trans-type diastereoselective synthesis of a wide variety of silylcyclopropanes bearing a quaternary carbon-stereocenter with good diastereo- and enantioselectivities (up to >19 : 1 dr and 97% ee). The preliminary experimental results of mechanistic studies showed that the steric repulsion between the TADDOL-derived phosphoramidite ligand and substrate would be an important factor, allowing access to different and reversed diastereospecific hydrosilylation in comparison to rhodium catalysis.
 Please wait while we load your content...
Please wait while we load your content...

