
RNA-cleaving DNAzymes for accurate biosensing and gene therapy

Abstract
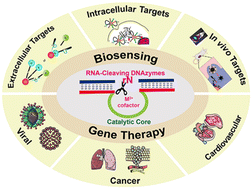
RNA-cleaving DNAzymes, which are single-stranded catalytic DNA, have attracted considerable attention in bioanalysis and biomedical applications because of their high stability, high catalytic activity, easy synthesis, easy functionalization, and modification. By incorporating these DNAzymes with amplification systems, the sensing platforms can be used to detect a series of targets with high sensitivity and selectivity. In addition, these DNAyzmes possess therapeutic potential by cutting the mRNA in cells and viruses to regulate the expression of the corresponding proteins. This review systematically summarizes the applications of RNA-cleaving DNAzymes in recent years, explaining the uniqueness and superiority of RNA-cleaving DNAzymes in biosensing and gene therapy. Finally, this review extends the discussion to the challenges and perspectives of applying RNA-cleaving DNAzyme as a diagnostic and therapeutic agent. This review provides the researchers with valuable suggestions and promotes the development of DNAzymes for accurate analysis, early diagnosis, and effective therapy in medicine and their broader applications beyond biomedicine.
- This article is part of the themed collections: Celebrating the 20th anniversary of the National Center for Nanoscience and Technology (NCNST) and Recent Review Articles
 Please wait while we load your content...
Please wait while we load your content...

