
Recent advances of structural/interfacial engineering for Na metal anode protection in liquid/solid-state electrolytes†
 *a
Yong
Zhao
*a
*a
Yong
Zhao
*a
Abstract
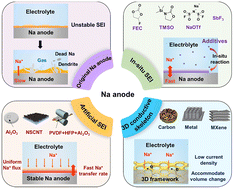
Na metal anode is one of most promising anode materials for next-generation secondary batteries. However, the practical application of Na anode is limited by dendritic growth, rapid volume change, and serious interface problems in the process of Na electroplating/stripping, resulting in low coulombic efficiency, short life, and safety issues of sodium metal batteries (SMBs). Herein, the cyclic instability mechanisms of the Na anode and the corresponding advanced protection strategies including in situ solid-electrolyte-interphase (SEI), artificial SEI, and three-dimensional conductive frame, are systematically reviewed. Notably, this review summarizes the latest research progress on interface modification and electrode modification of all-solid-state SMBs. Finally, the outlooks of anode interphase in SMBs are summarized and prospected, providing a promising way for high-energy and safe SMBs.
- This article is part of the themed collections: Celebrating 25 years of the Key Laboratory for Special Functional Materials at Henan University and Recent Review Articles
 Please wait while we load your content...
Please wait while we load your content...

