
Exploring the dynamics of DNA nucleotides in graphene/h-BN nanopores: insights from ab initio molecular dynamics†

Abstract
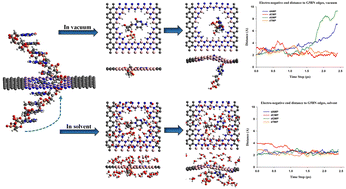
Nanopore devices based on graphene and h-BN heterostructures show outstanding electrical and physical characteristics for high throughput label-free DNA sequencing. On top of their suitability for DNA sequencing with the ionic current method, G/h-BN nanostructures are promising for DNA sequencing by employing the in-plane electronic current. The influence of the nucleotide/device interaction on the in-plane current has been widely explored for static-optimized geometries. Therefore, it is essential to investigate the dynamics of the nucleotides within the G/h-BN nanopores to gain a comprehensive view of their interaction with the nanopores. In this study, we investigated the dynamic interaction between nucleotides and nanopores in horizontal graphene/h-BN/graphene heterostructures. The insulating h-BN layer, where the nanopores are implemented, changes the in-plane charge transport mechanism into the quantum mechanical tunneling regime. We employed the Car-Parrinello molecular dynamics (CPMD) formalism to investigate the interaction of the nucleotides with nanopores in a vacuum as well as in an aqueous environment. The simulation was performed in the NVE canonical ensemble with an initial temperature of 300 K. The results indicate that the interaction between the electronegative ends of the nucleotides and the atoms at the nanopore edge is essential for the dynamic behavior of the nucleotides. Moreover, water molecules have a substantial effect on the dynamics and interactions of the nucleotides with nanopores.
- This article is part of the themed collection: Emerging concepts in nucleic acids: structures, functions and applications
 Please wait while we load your content...
Please wait while we load your content...

