
DNA–Au (111) interactions and transverse charge transport properties for DNA-based electronic devices†

Abstract
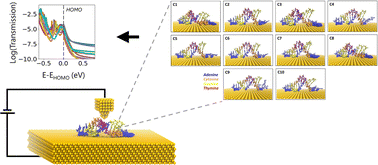
DNA's charge transfer and self-assembly characteristics have made it a hallmark of molecular electronics for the past two decades. A fast and efficient charge transfer mechanism with programmable properties using DNA nanostructures is required for DNA-based nanoelectronic applications and devices. The ability to integrate DNA with inorganic substrates becomes critical in this process. Such integrations may affect the conformation of DNA, altering its charge transport properties. Thus, using molecular dynamics simulations and first-principles calculations in conjunction with Green's function approach, we explore the impact of the Au (111) substrate on the conformation of DNA and analyze its effect on the charge transport. Our results indicate that DNA sequence, leading to its molecular conformation on the Au substrate, is critical to engineer charge transport properties. We demonstrate that DNA fluctuates on a gold substrate, sampling various distinct conformations over time. The energy levels, spatial locations of molecular orbitals and the DNA/Au contact atoms can differ between these distinct conformations. Depending on the sequence, at the HOMO, the charge transmission differs up to 60 times between the top ten conformations. We demonstrate that the relative positions of the nucleobases are critical in determining the conformations and the coupling between orbitals. We anticipate that these results can be extended to other inorganic surfaces and pave the way for understanding DNA–inorganic interface interactions for future DNA-based electronic device applications.
- This article is part of the themed collection: Emerging concepts in nucleic acids: structures, functions and applications
 Please wait while we load your content...
Please wait while we load your content...

