
Mechanistic insight into the structure, thermodynamics and dynamics of equilibrium gels of multi-armed DNA nanostars

Abstract
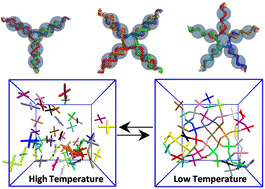
The unique sequence specificity rule of DNA makes it an ideal molecular building block for constructing periodic arrays and devices with nanoscale accuracy and precision. Here, we present the self-assembly of DNA nanostars having three, four and five arms into a gel phase using a simplistic coarse-grained bead-spring model developed by Z. Xing, C. Ness, D. Frenkel and E. Eiser (Macromolecules, 2019, 52, 504–512). Our simulations show that the DNA nanostars form a thermodynamically stable fully bonded gel phase from an unstructured liquid phase with the lowering of temperature. We characterize the phase transition by calculating several structural features such as the radial distribution function and structure factor. The thermodynamics of gelation is quantified by the potential energy and translational pair-entropy of the system. The phase transition from an arrested gel phase to an unstructured liquid phase has been modelled using a two-state theoretical model. We find that this transition is enthalpy driven, and loss of configuration and translational entropy is counterpoised by enthalpic interaction of the DNA sticky-ends, which gives rise to a gel phase at low temperature. The absolute rotational and translational entropy of the systems, measured using a two-phase thermodynamic model, also substantiates the gel transition. The slowing down of the dynamics upon approaching the transition temperature from a high temperature demonstrates the phase transition to a gel phase. A detailed numerical simulation study of the morphology, dynamics and thermodynamics of DNA gelation can provide guidance for future experiments, is easily extensible to other polymeric systems, and is expected to help in understanding the physics of self-assembly.
- This article is part of the themed collection: Emerging concepts in nucleic acids: structures, functions and applications
 Please wait while we load your content...
Please wait while we load your content...

