
Rationalization and tuning of doublet emission in organic radicals†
 *a
and
David
Casanova
ab
*a
and
David
Casanova
ab
Abstract
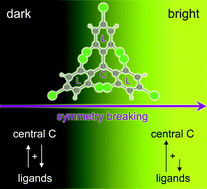
Neutral organic radical emitters are promising alternative candidates for efficient OLEDs. Their potential relies on their ability to avoid spin statistics loss of charge recombination as in closed shell emitters. Yet, these radicals generally present limited luminescence that was shown to be enhanced via chemical functionalization. Here we provide a detailed physical interpretation of the origin of the intrinsic poor emissive properties of TTM, a paradigmatic doublet emitter, from a computational perspective. The weak emission is rationalized on the basis of the mixing of intramolecular charge transfer (CT) excitations between the radical center and the ligands. We demonstrate that significant cancellation of the transition dipole moments of these CT contributions results in low oscillator strength and show how the modulation of their relative contributions to the composition of the lowest excited doublet state via donor substitution can brighten the organic radical. We illustrate this analysis through the study of a series of TTM derivatives. This study contributes to further the understanding of the photophysics of organic radicals and provides new insights for the tuning of their luminescence by rational design.
- This article is part of the themed collections: 2022 Journal of Materials Chemistry C Most Popular Articles and Journal of Materials Chemistry C Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

