
Triphenylene-ethylammonium tetrachlorometallate salts: multicolumnar mesophases, thermochromism and Langmuir films†‡
 a
María Jesús
Baena,
a
Bertrand
Donnio,
*b
Benoît
Heinrich,
b
Lucía
Gutiérrez,
c
Silverio
Coco
*a
and
Pablo
Espinet
*a
a
María Jesús
Baena,
a
Bertrand
Donnio,
*b
Benoît
Heinrich,
b
Lucía
Gutiérrez,
c
Silverio
Coco
*a
and
Pablo
Espinet
*a
Abstract
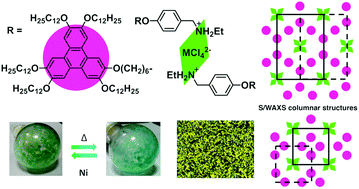
This study reports new mesomorphic benzylethylammonium salts containing a 2,3,6,7,10-pentakis(dodecyloxy)-11-(hexyloxy)triphenylene unit and Cl− or [MCl4]2− (M = Cu, Ni, Co, Mn) as counterions. They show a rich columnar polymesomorphism, characterized by polarized optical microscopy (POM), differential scanning calorimetry (DSC), and small- and wide-angle X-ray scattering (S/WAXS). All the compounds self-organize into multisegregated structures in the solid state and in the mesophases, with the alternation within large superlattices of triphenylene-containing columns and benzylammonium chloride or tetrachorometallate fragments. Interestingly, the nickel derivative displays a continuous and reversible thermochromic behavior in the range of 50–80 °C. Magnetic measurements and UV-Vis data at different temperatures, support changes in the coordination sphere of the Ni(II) atom from octahedral to tetrahedral. Furthermore, these systems form stable and homogeneous Langmuir films at the air–water interface. These films have been successfully transferred onto solid substrates giving rise to nanostructured LB films with a clearly defined bilayer structure in which each layer consists of molecular columns aligned along the surface.
- This article is part of the themed collection: Editor’s Choice: Advances and New Avenues in Liquid Crystal Science
 Please wait while we load your content...
Please wait while we load your content...

