
Manipulating Förster and Dexter interactions between a thermally activated delayed fluorescence host and a phosphorescent dopant for highly efficient solution-processed red and white OLEDs†

Abstract
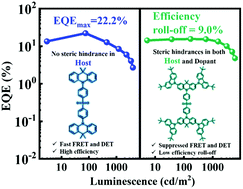
Solution-processed phosphorescent organic light-emitting diodes (PhOLEDs) have been known as promising alternatives to their vacuum deposited counterparts due to their excellent cost-effectiveness. However, the solution-processed devices still suffer from low efficiencies especially for the saturated red emission. Herein, we designed and demonstrated very efficient solution-processed red PhOLEDs by introducing a thermally activated delayed fluorescence (TADF) host with and without inert peripheral units, i.e., 4CzDMAC-DPS and DMAC-DPS respectively, to manipulate Förster and Dexter interactions. Compared with the steric hindrance on both the host and guest, Förster and Dexter energy transfers were simultaneously promoted when removing the hindrance units on the TADF host. This is mainly attributed to the larger radius of Förster energy transfer and closer intermolecular distance between DMAC-DPS and the guest, compared with those of the 4CzDMAC-DPS and guest system. As a result, the state-of-the-art solution-processed saturated red PhOLEDs achieved a high maximum external quantum efficiency (EQE) of 22.2% with an emission peak beyond 610 nm. In contrast, the TADF host with the additional hindrance units played an important role in achieving a low efficiency roll-off (< 10% at 1000 cd m−2) without sacrificing the EQE. Moreover, ternary-blended solution-processed PhOLEDs exhibited easy color tuning from red to white, which was dominated by the energy transfer radius mediated by the hindrance effect of the TADF compound.
- This article is part of the themed collection: Materials for thermally activated delayed fluorescence and/or triplet fusion upconversion
 Please wait while we load your content...
Please wait while we load your content...

