
Near-infrared-to-visible upconversion from 980 nm excitation band by binary solid of PbS quantum dot with directly attached emitter†

Abstract
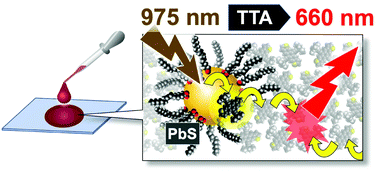
Long-wavelength NIR photon upconversion (UC) by triplet–triplet annihilation in cast solid is reported. NIR excitation at 975 nm of the binary solid of PbS quantum dot (QD) and an anthradithiophene derivative, which can attach to the QD directly, gave a UC quantum yield of 0.2% (vs. 50% full-scale) and threshold intensity of 2.5 W cm−2. Triplet energy transfer from QD was found to be accelerated by the direct attachment, based on the UC emission dynamics.
- This article is part of the themed collection: Materials for thermally activated delayed fluorescence and/or triplet fusion upconversion
 Please wait while we load your content...
Please wait while we load your content...

