
Recent progress in pyrazinacenes containing nonbenzenoid rings: synthesis, properties and applications
 a
Qichun
Zhang
*ab
a
Qichun
Zhang
*ab
Abstract
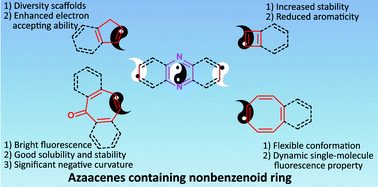
Azaacenes have emerged as a new and important class of organic materials, and their synthesis strategies and applications as organic semiconductors have gained significant progress in recent years. Generally, adopting sterically-shielding substituents such as attaching large silylethynyl groups at selected peripheral positions or extending the π-conjugation with pyrene units is the common method used to stabilize larger azaacenes. However, another way to stabilize and enlarge azaacenes, as well as to tune their optical and electronic properties by inserting nonbenzenoid rings such as four-membered rings into the skeletons, has also been developed but has received much less attention. Therefore, in this review, we summarize the recent progress in their syntheses, properties, and applications in organic electronics. Moreover, we highlight the effect of nonbenzenoid units in the systems. Finally, we discuss the current challenges and perspectives through comparison with conventional azaacenes.
- This article is part of the themed collection: Special issue in honour of Daoben Zhu
 Please wait while we load your content...
Please wait while we load your content...

