
A two-dimensional polymer memristor based on conformational changes with tunable resistive switching behaviours†
 *a
Xi
Yu
*a
and
*a
Xi
Yu
*a
and
Abstract
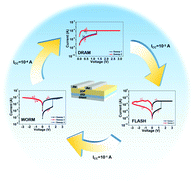
In this work, through rational monomer design we integrated the conformational change mechanism into a two-dimensional covalent polymer and implemented 3 different low-power memory behaviors on the same device by tuning the compliance current (ICC), including nonvolatile write-once-read-many-times (WORM) memory behaviour, nonvolatile FLASH memory behaviour and volatile dynamic random access memory (DRAM) behaviour. Furthermore, 2DP-based memory devices exhibit excellent flexibility and thermal stability, which is a desirable property for applications in wearable electronics and a high temperature environment. Finally, the conformation-induced conductance switching under an electric field was confirmed by UV-vis spectra. The differences in memory behaviour were attributed to the inherent differences in the degree of regioregularity and ease of the conformational relaxation of the field-induced rearrangement of carbazole groups.
- This article is part of the themed collection: Special issue in honour of Daoben Zhu
 Please wait while we load your content...
Please wait while we load your content...

