
Axially chiral bay-tetraarylated perylene bisimide dyes as non-fullerene acceptors in organic solar cells†‡
 *ab
*ab
Abstract
A series of twisted perylene bisimide (PBI) dyes bearing different imide substituents as well as various aryl substituents in 1,6,7,12 bay positions inducing conformationally stable axial chirality are investigated as non-fullerene acceptors (NFAs) in organic solar cells (OSCs). In a comprehensive study, both imide and aryl substituent variations are tested within a simple inverted bulk heterojunction (BHJ) architecture with donor polymer PCE-10. The results are rationalized by single crystal structure analyses revealing the importance of the sterical demand imparted by the substituents in both positions on the packing arrangements. Comparative studies of racemic and enantiopure samples of these axially chiral dyes showed that the OSC performance is only affected by the enantiopurity if the chromophore is not well shielded. In that case, intimate π–π-stacking is disfavored for enantiopure compounds, thereby improving the OSC efficiency significantly by up to 24%. For a more shielded PBI bearing bulky 2,6-diisopropylphenyl (Dipp) substituents at imide and 2-naphthyl at bay positions, a maximum power conversion efficiency (PCEmax) of 4.3% is achieved even for the racemate.
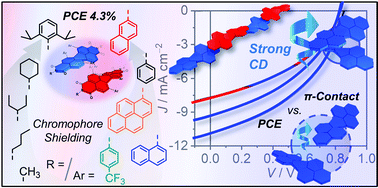
- This article is part of the themed collection: Special issue in honour of Daoben Zhu
 Please wait while we load your content...
Please wait while we load your content...

