
Recent advances in biomedical applications of bacterial outer membrane vesicles
Liang
Han  *a
*a
*a
Abstract
Nanoscale and non-self-replicating outer membrane vesicles (OMVs) are naturally secreted by some bacteria with their structures and compositions similar to that of the outer membrane of parental bacteria. With some specific bacterial physiological characteristics, e.g., immunomodulations and intercellular communications, OMVs have been intensively studied and extensively used and engineered as vaccines, immunotherapeutic drugs, anti-bacterial agents, and drug delivery carriers. In this review, we describe the extraction, characterization, and functionalization of OMVs with emphasis on the latest progress and prospects in biomedical applications.
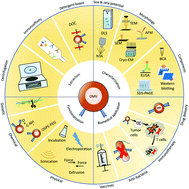
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

