
Thioether-based ROS responsive polymers for biomedical applications
 *a
and
David
Mecerreyes
ab
*a
and
David
Mecerreyes
ab
Abstract
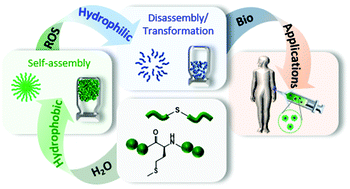
Reactive oxygen species (ROS) play a key role in several biological functions of living organisms such as regulation of cell signalling, production of some hormones, modulation of protein function or mediation of inflammation. In this regard, ROS responsive polymers are ideal candidates for the development of stimuli-responsive biomaterials for target therapies. Among different ROS-responsive polymers, those containing thioether groups are widely investigated in the biomedical field due to their hydrophobic to hydrophilic phase transition under oxidative conditions. This feature makes them able to self-assemble in aqueous solutions forming micellar-type nanoparticles or hydrogels to be mainly used as drug carriers for local therapies in damaged body areas characterized by high ROS production. This review article collects the main findings about the synthesis of thioether-based ROS responsive polymers and polypeptides, their self-assembly properties and ROS responsive behaviour for use as injectable nanoparticles or hydrogels. Afterward, the foremost applications of the thioether-based ROS responsive nanoparticles and hydrogels in the biomedical field, where cancer therapies are a key objective, will be discussed.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

