
Recent progress of nanomedicine in secreted phospholipase A2 as a potential therapeutic target
 a
and
Lesan
Yan
*a
a
and
Lesan
Yan
*a
Abstract
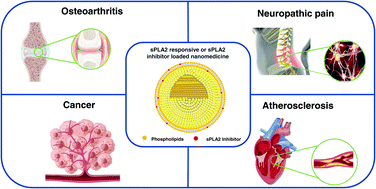
Overexpressed secretory phospholipase A2 (sPLA2) is found in many inflammatory diseases and various types of cancer. sPLA2 can catalyze the hydrolysis of phospholipid sn-2 ester bonds to lysophosphatidylcholine and free fatty acids, and its catalytic substrate and downstream products mediate a series of cascade reactions and inflammatory responses. Furthermore, different subtypes of sPLA2 can participate in different physiological processes by driving unique lipid pathways. Recently, many diseases have not been treated by appropriate chemotherapy methods due to low bioavailability and severe side effects of clinically available small-molecule drugs. Therefore, they have great development prospects of revealing the therapeutic mechanism of sPLA2 and use sPLA2 as a potential therapeutic target for designing and exploring new drugs and their delivery systems. Notably, the emergence of nanomedicines in recent years provides a practical and innovative means for overcoming the challenges associated with chemotherapy. With these considerations in mind, this paper systematically reviews recent studies on nanomedicines targeting sPLA2 overexpression in various diseases during the past few years.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

